
2011
Magnus Rueping, Teerawut Bootwicha, Hannah Baars and Erli Sugiono
Beilstein J. Org. Chem. 2011, 7, 1680–1687

ABSTRACT:
A simple, practical and efficient continuous-flow hydration–condensation protocol was developed for the synthesis of α,β-unsaturated ketones starting from alkynes and aldehydes by employing a heterogeneous catalyst in a flow microwave. The procedure presents a straightforward and convenient access to valuable differently substituted chalcones and can be applied on multigram scale. We have developed a general protocol to access a series of valuable differently substituted chalcones. Starting from commercially available alkynes and aldehydes, a continuous-flow hydration–condensation protocol leads to the desired products in good to excellent yields. The reactions were sequentially introduced into the flow cell and performed several times without the heterogeneous catalyst needing to be changed, demonstrating the high robustness of this catalytic system. Additionally, this new method was readily applied for the preparation of chalcones in multigram quantities. The technology presented is advantageous over classical non-microwave batch reactions in particular with regard to the continuous harvesting of the product, the fast optimization of the reaction parameters, the simple operation and reliability, and the restriction of byproduct formation, especially the formation of methyl ketones and propargylic alcohols.
Magnus Rueping, Chandra M. R. Volla, Michael Bolte, Gerhard Raabe
Adv. Synth. Catal. 2011, 353, 2853 - 2859

ABSTRACT:
An asymmetric organocatalyzed reaction sequence involving a Michael addition of various 1,3-dicarbonyl compounds to α,β-unsaturated aldehydes with subsequent diastereoselective Pictet–Spengler cyclization has been developed. The substrate scope was found to be general and optically active indoloquinolizidines were isolated as single diastereomers in high yields with high to excellent enantioselectivites. In addition to tryptamine, the reaction has also been successfully applied to other nucleophiles including o-aminobenzylamine and anthranilamide giving rise to pyridoquinauolines and quinazolinones.
Magnus Rueping, Lukas Hubener
Synlett 2011, 1243 - 1246
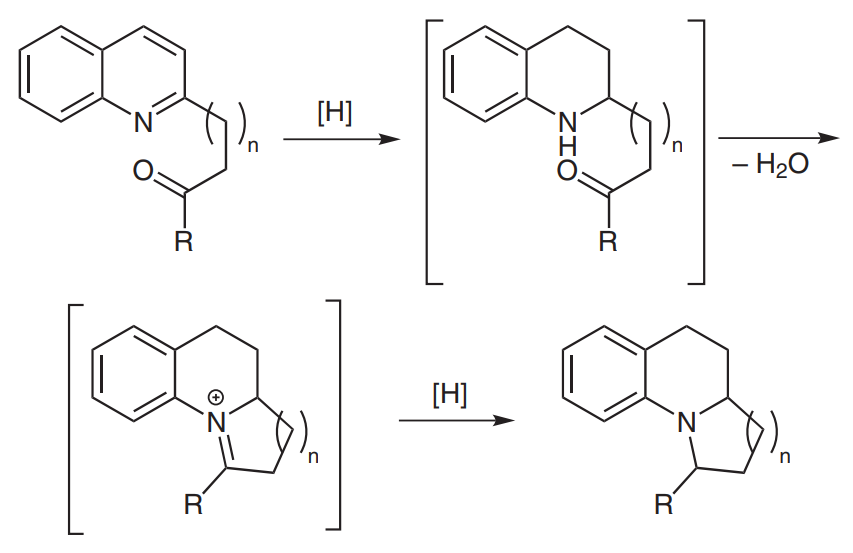
ABSTRACT:
A catalytic enantioselective synthesis of a new class of quinolizidines and indolizidines is presented. An asymmetric Brønsted acid catalyzed hydrogenation cascade as well as a sequential Brønsted acid/metal catalyzed hydrogenation protocol of 2-substituted quinolines yields benzofused quinolizidines and indolizidines in good yields with high diastereo- and enantioselectivities.
Magnus Rueping, Rene M. Koenigs, Ruediger Borrmann, Jochen Zoller, Thomas E. Weirich, Joachim Mayer
Chem. Mater. 2011, 23, 2008 - 2010

ABSTRACT:
The synthesis of iridium nanoparticles on both functionalized and unfunctionalized carbon nanotubes, using for the first time a stabilizer-free, hydrogenolytic approach for nanoparticle formation was reported. Both the temperature of the hydrogenolysis and the oxidative pretreatment play important roles in the preparation of the nanoparticles that are formed in a nucleation-limited pathway. Although at low temperature nucleation is slow and large particles are formed, the use of high temperatures during the hydrogenolysis process favors the formation of highly reactive iridium nanoparticles. The Ir@CNT catalyst formed by this method not only exhibits the highest catalytic activity in batch reduction of N-heterocycles to date but it can additionally be employed in continuous flow reductions. Further research is directed to a better understanding of the nanoparticle formation employing different metal precursors as well as further investigations toward applications in catalysis.
Magnus Rueping, Uxue Uria, Ming-Yuan Lin, Iuliana Atodiresei
J. Am. Chem. Soc. 2011, 133, 3732-3735

ABSTRACT:
Chiral contact ion-pair catalysis with particular focus on metal-free processes is gaining in interest. As a result, new perspectives are opened, and highly stereoselective transformations, traditionally performed under metal catalysis, can be realized. Herein, we report the development of an unprecedented asymmetric Brønsted acid-catalyzed allylic alkylation. The concept relies on chiral contact ion-pair catalysis, in which the chiral organic counteranion of an allylic carbocation induces high enantioselectivities and allows access to biologically relevant chromenes in good yields and with excellent enantioselection. These results highlight the potential of Brønsted acids in promoting highly stereoselective allylic reactions via a chiral contact ion pair. The present approach is particularly attractive as it avoids the use of often toxic metals and sensitive intermediates. We are thus confident that the concept of asymmetric ion-pair catalysis in which the chiral information is efficiently transferred from the Brønsted acid anions to the product will find broader application in reactions involving carbocationic intermediates
Magnus Rueping, Sadiya Raja, Alberto Núñez
Adv. Synth. Catal. 2011, 353, 563 - 568

ABSTRACT:
A new enantioselective Brønsted acid-catalyzed Friedel–Crafts reaction of indole with cyclic imines has been develeoped. This organocatalytic reaction provides for the first time optically active indolindolinone derivatives in high yields and with excellent enantioselectivities (up to 91% ee) under mild reaction conditions. The asymmetric Brønsted acid-catalyzed Friedel–Crafts reaction of differently substituted indoles with cylic imines is highly efficien and selective. This method represents the first enantioselective aza-Friedel–Crafts reaction with cyclic imines as electrophiles and provides the corresponding optically active indolindolinone derivatives in high yields, with high enantioselectivities and broad functional group tolerance. The products are attractive, as they possess an indole and an indolin-3-one core, which are both suitable for further derivatization.
17. Enantioselective Organocatalytic Synthesis of Quaternary α-Amino Acids Bearing a CF3 Moiety
Ralph Husmann, Erli Sugiono, Stefanie Mersmann, Gerhard Raabe, Magnus Rueping, Carsten Bolm
Org. Lett. 2011, 13, 1044-1047
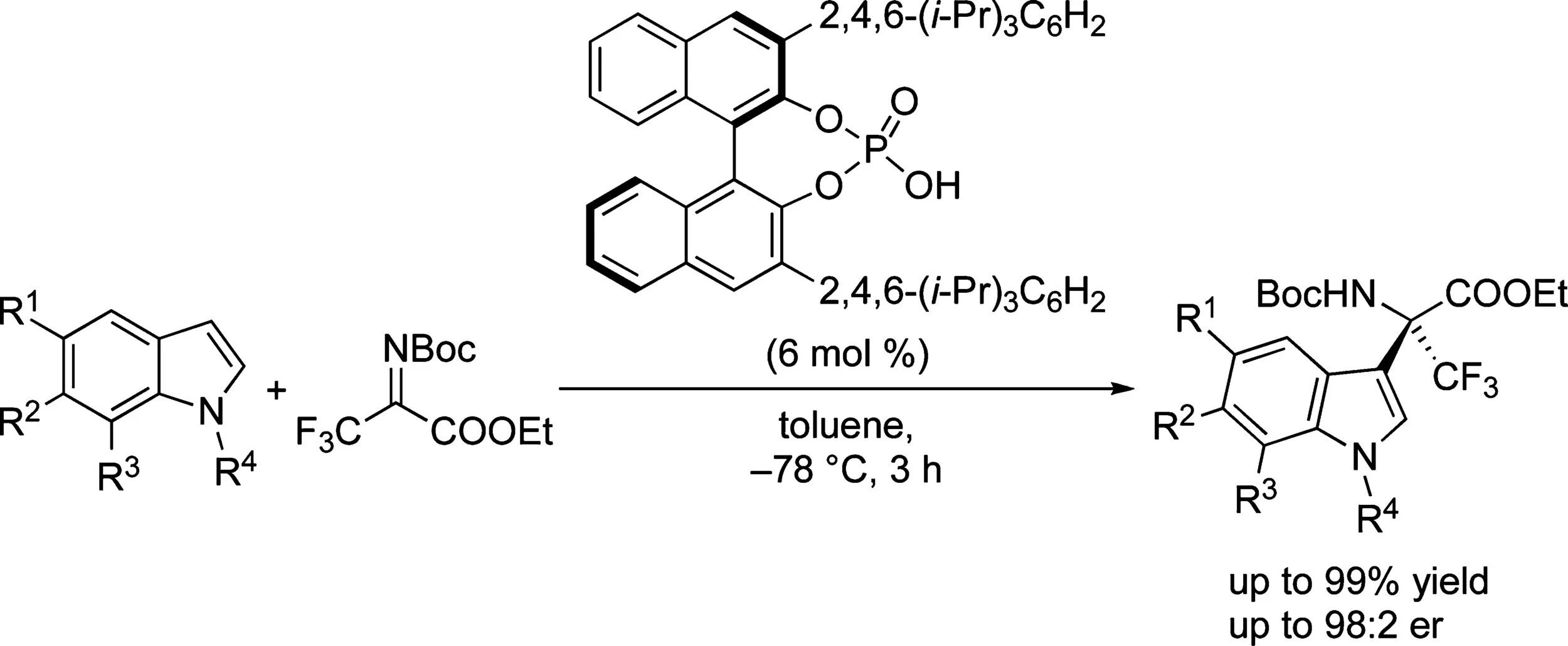
ABSTRACT:
A highly enantioselective Friedel−Crafts reaction catalyzed by a chiral phosphoric acid was developed. N-Boc-protected ethyl trifluoropyruvate imine was activated by 6 mol % of catalyst and reacted with a wide variety of indole derivatives to afford quaternary α-amino acids in excellent yields (up to 99%) and high enantioselectivities (up to 98:2 er). The N-Boc-protected trifluoropyruvate-derived imine proved most effective in terms of yield and stereoselectivity. A wide variety of substituents in the 5-, 6-, and 7-positions of the indole were tolerated. The absolute configuration of a representative product was determined by comparison of calculated and experimental ECD spectra. Deprotection of the Boc-derivative was accomplished giving the amino acid ester in excellent yield while retaining the stereochemical information.
16. Copper Catalyzed C−H Functionalization for Direct Mannich Reactions
Magnus Rueping, Nikita Tolstoluzhsky
Org. Lett. 2011, 13, 1095-1097

ABSTRACT:
A protocol for a practical and direct addition of α- and γ-alkyl azaarenes to N-sulfonyl aldimines has been developed. Copper salts act as efficient Lewis acid catalysts for direct Mannich-type reactions providing a mild and fast access to various functionalized heterocycles representing an efficient and atom-economical protocol for the direct α- and γ-addition of 2- and 4-alkyl azaarenes to aldimines. The reaction proceeds through a copper catalyzed direct C−H bond functionalization and provides a set of different heterocycle-containing amines in good yields.
Magnus Rueping, Teerawut Bootwicha, Erli Sugiono
Synlett 2011, 323-326

ABSTRACT:
We have developed an asymmetric calcium-catalyzed direct Mannich reaction of cyclic 1,3-dicarbonyl compounds with different N-Boc-protected aldimines. The chiral calcium catalysts are derived from BINOL phosphoric acids which have also shown to be catalysts for this reaction. In addition to existing methods, we were able to demonstrate that carbonyl donors, such as cyclohexadione as well as pyrone, can be applied in asymmetric Mannich reactions and that the corresponding products, which are valuable intermediates in organic synthesis, can be obtained with good enantiomeric excess.
Magnus Rueping and Chandra M. R. Volla
RSC Adv., 2011, 1, 79-82

ABSTRACT:
A highly diastereoselective methodology for the efficient synthesis of functionalized indolo[2,3-a]quinolizidine skeletons in a one-pot operation has been developed. The protocol makes use of simple and inexpensive starting materials such as tryptamines, 1,3-dicarbonyl compounds and α,β-unsaturated aldehydes in the presence of a Brønsted acid catalyst. We have developed a highly efficient and diastereoselective one-pot reaction of tryptamine with 1,3-dicarbonyl compounds and unsaturated aldehydes. Notably, in the course of the reaction, four new bonds are constructed of which two are C–C bonds generating chiral centers. Several unsaturated aldehydes have been employed and afforded the corresponding products in good yields and high diastereoselectivities. The usefulness of this method is certified by its efficiency, easy operability, mild reaction conditions, cheap starting materials as well as excellent diastereoselectivity.
13. Brønsted Acid Catalysis: Hydrogen Bonding versus Ion Pairing in Imine Activation
Matthias Fleischmann, Diana Drettwan, Erli Sugiono, Magnus Rueping, Ruth M. Gschwind
Angew. Chem. Int. Ed. 2011, 50, 6364-6369

ABSTRACT:
It was shown that NMR spectroscopy is the method of choice to clearly distinguish between the activation modes of hydrogen bonding and ion pairing in Brønsted acid catalysis. Before this study, it was assumed that full protonation of the imine resulted in the formation of an ion pair, which would subsequently react with a nucleophile. However, our experiments clearly show that besides ion pairing, hydrogen bonding exists. The relative hydrogen-bond strength in OH(1⋅XOH⋅⋅⋅N) (2>3>4) and the relative amount of OH(1⋅XOH⋅⋅⋅N) at room temperature (4>3≈2) show that both hydrogen-bond strength and the amount of the OH species are decisive for the reaction. Furthermore, the ratio between hydrogen bonding and ion pairing (OH, NH) can be manipulated readily by simply introducing substituents with different electronic properties. These results provide insight into the different activation modes in Brønsted acid catalysis and are expected to guide the development of more efficient catalytic systems.
12. Modulating the Acidity: Highly Acidic Brønsted Acids in Asymmetric Catalysis
Magnus Rueping, Boris J. Nachtsheim, Winai Ieawsuwan, Iuliana Atodiresei
Angew. Chem. Int. Ed. 2011, 50, 6706-6720

ABSTRACT:
Recently, chiral highly acidic Brønsted acids have emerged as powerful catalysts for enantioselective CC and CX bond-forming reactions. Their strong acidity renders them valuable tools for the activation of imines, carbonyl compounds, and other weakly basic substrates. As a result, new perspectives are opened and highly stereoselective transformations based on the concept of chiral contact-ion-pair catalysis can be realized. This Minireview gives an overview of the design and application of these new organocatalysts and presents recent results in this rapidly growing field.
Magnus Rueping and René M. Koenigs
Chem. Commun. 2011, 47, 304-306

ABSTRACT:
The first Brønsted acid differentiated metal catalyzed hydrogenation by kinetic discrimination was reported. Applying this concept, it was shown for the first time, that skillful combination of chiral N-triflylphosphoramides and cheap, racemic iridium complexes can be used to obtain good enantioselectivities. Furthermore, the optimal catalyst combination can be readily determined by utilizing a fast combinatorial approach in which racemic metal complexes are simply combined with chiral acids in order to identify the ideal catalyst system. To date, only chiral phosphoric aciddiesters have been utilized in combination with metal complexes. Based on the different coordination properties of N-triflylphosphoramides the repertoire of combined Brønsted acid–metal catalysis is not only enhanced by an additional Brønsted acid, but also by the possibility of designing substantially different chiral complexes bearing mono- or bidentate or even non-coordinating chiral anions that could conceivably exhibit distinct activation modes.
10. Dual catalysis: combining photoredox and Lewis base catalysis for direct Mannich reactions
Magnus Rueping, Carlos Vila, René M. Koenigs, Konstantin Poscharny and David C. Fabry
Chem. Commun. 2011, 47, 2360-2362

ABSTRACT:
A dual catalytic system combining photoredox and Lewis base catalysis has been developed. By the appropriate choice of light source and catalyst, the photoredox cycle can be optimally modulated to match the base catalyzed reaction cycle to provide the corresponding products under mild reaction conditions. We have developed a dual catalytic system for the Mannich reaction by combining photoredox catalysis and Lewis base catalysis. The reaction can effectively be conducted with only 1 mol% photocatalyst loading. The perfect interplay of both catalytic cycles is crucial for obtaining good yields, as the highly reactive intermediates can undergo undesirable pathways yielding complex reaction mixtures. However, we were able to demonstrate that the photocatalytic cycle can be adapted by carefully fine tuning the light source and the photocatalyst. Most importantly the latter adjustments have to be carried out in the presence of the Lewis base catalyst. The dual catalytic process described demonstrates a further advancement in the recent area of light driven strategies in which the choice of the appropriate light source is the key to induce an efficient and selective transformation.
Magnus Rueping, KyoungLang Haack, Winai Ieawsuwan, Henrik Sundén, Magda Blanco and Fenja R. Schoepke
Chem. Commun. 2011, 47, 3828-3830

ABSTRACT:
The design of biologically inspired, multi-component cascade reactions enables the targeted synthesis of assorted structurally complex products. Similar to regulation in cells the reaction path is controlled by the substrate concentration and complex enantiopure products with high structural diversity are provided. To the best of our knowledge, the quadruple reaction cascade that we have developed is the first example of a complex asymmetric cascade reaction in which the course of the reaction can be controlled by merely changing the substrate concentration. Similar to enzymatic processes in the metabolism, it is possible to selectively synthesize the quadruple product or the domino product by simply increasing or decreasing the aldehyde concentration. With regard to the mechanism, the quadruple reaction introduced here is an example of a Lewis base catalyzed iminium–enamine–iminium–enamine activation sequence. Thus, we have developed a substrate regulated, asymmetric, metal-free reaction in which the reaction pathway can be controlled via the concentration. Depending on the concentration of the individual substrates, targeted selection between a double and a quadruple reaction can be made. The products of both reaction cascades were isolated for a wide range of substrates in good to very good yields and with excellent enantioselection. With the aid of this first example of chemoselective reaction control through substrate concentration it is possible, with resourceful reaction planning, to access numerous complex products with high functional diversity and good stereoselectivity, using simple educts in only a few reaction steps as is often seen in nature. This is an example of synthetic chemistry mimicking nature in its precise reaction control. Therefore, the reaction presented above is bound to serve as a template for further studies of efficient reaction control through the substrate concentration.
Magnus Rueping, Shaoqun Zhu and René M. Koenigs
Chem. Commun. 2011, 47, 8679-8681

ABSTRACT:
A visible light mediated, carbon–phosphorus bond forming reaction has been developed. With the use of a readily available photoredox catalyst, α-amino phosphonates were obtained in good yields under mild reaction conditions. We have developed the first protocol for an oxidative carbon–phosphorus bond forming reaction using a visible light photoredox catalysis procedure. The generation of highly reactive iminium ions under ambient conditions can be achieved in a biphasic mixture of toluene and water, where the formation of the typically observed amide by-product through over-oxidation is suppressed. This effect can be rationalized by the interception of the peroxy species by water as well as the beneficial effect of phase separation. This observation will be useful for further reaction development in photoredox catalysis and visible light mediated aerobic oxidations. The new visible light mediated oxidative phosphonylation enables ready access to valuable α-amino phosphonates, which have been described as α-amino acid mimics. Attractive features of this protocol are the readily available starting materials, the operational simplicity and practicability as well as the mild reaction conditions using visible light as the energy source.
7. Visible light mediated azomethine ylide formation—photoredox catalyzed [3+2] cycloadditions
Magnus Rueping, Daniele Leonori and Thomas Poisson
Chem. Commun. 2011, 47, 9615-9617

ABSTRACT:
The synthesis of highly functionalised N-heterocycles has been achieved by the visible light mediated photoredox conversion of tertiary amines to azomethine ylides and their further reaction with maleimide derivatives as dipolarophiles. We have reported a new visible light mediated photoredox transformation of tertiary amines which leads to the formation of azomethine ylides. For the first time, a photoredox cycle has been merged with a cycloaddition reaction,8 resulting in the simultaneous and diastereoselective formation of two C–C single bonds. The high complexity of the product formed under these mild reaction conditions makes this new method attractive for the formation of highly functionalised heterocycle
Magnus Rueping, Matthias Leiendecker, Arindam Das, Thomas Poisson and Lan Bui
Chem. Commun. 2011, 47, 10629-10631

ABSTRACT:
A transition metal-free Heck-type cyclization/isomerization reaction has been developed. Mediated by potassium tert-butoxide and phenanthroline a variety of benzofuran derivatives have been synthesized. We have developed a transition metal-free intramolecular Heck-type cyclization/isomerization reaction mediated by KOtBu. A variety of benzofuran derivatives are accessible through a reaction pathway which involves aryl and stabilized tertiary benzhydryl radicals. Due to the operational simplicity, the method is expected to find further application in the synthesis of heterocyclic compounds of interest.
Magnus Rueping and Winai Ieawsuwan
Chem. Commun. 2011, 47, 11450-11452

ABSTRACT:
A highly efficient Brønsted acid catalyzed enantioselective Nazarov cyclization–bromination reaction has been developed. The protocol gives access to highly functionalized trans-4,5-substituted 5-bromocyclopentenone derivatives in good yields and with excellent enantioselectivities. We have developed the first asymmetric Brønsted acid-catalyzed Nazarov cyclization–halogenation reaction. Two chiral centers, a tertiary and a quaternary one, have been established during this transformation. The present method provides, for the first time, a variety of α-brominated cyclopent-2-enones with a wide substrate scope and with excellent enantioselectivities.
4. Visible-light photoredox catalyzed oxidative Strecker reaction
Magnus Rueping, Shaoqun Zhu and René M. Koenigs
Chem. Commun. 2011, 47, 12709-12711

ABSTRACT:
An aerobic photocatalytic oxidative cyanation of tertiary amines providing valuable α-aminonitriles in good to excellent yields was developed. Mild reaction conditions and low catalyst loading are attractive features of the protocol. We have developed the first efficient aerobic, photocatalytic oxidative cyanation of tertiary amines employing cheap and readily available KCN as CN− source that yields valuable α-aminonitriles under mild reaction conditions and with low catalyst loadings. In addition, we could present that photoredox catalyzed oxidation reactions can be applied for the functionalization of acyclic aniline derivatives. Furthermore, an alternative pathway for the generation of the iminium ion intermediate was proposed. The generation of the carbon centred neutral radical intermediate suggested thereby can be further explored to design radical-type processes.
3. Chiral Brønsted acids in enantioselective carbonyl activations – activation modes and applications
Magnus Rueping, Alexander Kuenkel and Iuliana Atodiresei
Chem. Soc. Rev. 2011, 40, 4539-4549

ABSTRACT:
Chiral phosphoric acids and derivatives have attracted considerable attention as a powerful tool in asymmetric catalysis. Various enantioselective reactions have been developed by using these efficient Brønsted acid organocatalysts. Although initially the activation was restricted to reactive Brønsted basic substrates, recent reports are demonstrating the versatility of phosphoric acid catalysts in the activation of carbonyl compounds in a stereochemically controlled fashion. This tutorial review gives an overview of enantioselective Brønsted acid catalyzed transformations with the main focus on carbonyl activation. Different activation modes, key features of the catalysts and the applied substrates are presented and discussed with the goal to elucidate the origin of stereoselectivity in these Brønsted acid catalyzed transformations.
Magnus Rueping, Jeremy Dufour and Fenja R. Schoepke
Green Chem., 2011, 13, 1084-1105

ABSTRACT:
This review focuses on recent advances in catalytic metal-free transfer hydrogenations. In recent years dihydropyridines have been widely used as reducing agents in organocatalytic reductions. Analogous to nature's co-factor nicotinamide adenine dinucleotide (NADH), Hantzsch esters serve as efficient hydride donors. In combination with chiral organocatalysts, including chiral secondary amines, hydrogen bond donors or Brønsted acids, efficient catalytic asymmetric reductions have been developed which provide a diverse set of biologically active compounds, synthetic building blocks and natural products. These recent advances in developing green and sustainable reductions employing organocatalytic strategies are promising and important alternatives to conventional metal- and bio-catalyzed reductions.
Magnus Rueping, Thomas Theissmann, Mirjam Stoeckel and Andrey P. Antonchick
Org. Biomol. Chem. 2011, 9, 6844-6850

ABSTRACT:
A convenient protocol for the enantioselective synthesis of 4-substituted tetrahydroquinolines has been developed. Chiral BINOL phosphoric acids promote the reduction of a wide range of 4-substituted quinolines with Hantzsch esters with good to high levels of enantioselectivity. We have succeeded in developing the first asymmetric hydrogenation of biologically relevant 4-substituted quinolines. The protocol provides direct access to a large variety of 4-aryl- and 4-alkyl-substituted tetrahydroquinolines in good yields and good to high enantiomeric excesses. Further work will be devoted to the application of this methodology in the synthesis of more complex molecular architectures.