
2014
Roman Pluta, Magnus Rueping
Chem. Eur. J. 2014, 20, 17315-17318

ABSTRACT:
The chemoselective trifluoromethylthiolation of nitrogen nucleophiles and thiols using N-(trifluoromethylthio)phthalimide under mild, metal-free conditions is described. A series of trifluoromethanesulfenamides and unsymmetrical disulfides is prepared from the corresponding aliphatic and aromatic amines and thiols in good yields. The reactions are operationally simple and tolerate a wide variety of functional groups. Trifluoromethanesulfenamides and disulfides belong to interesting classes of organic molecules which possess remarkable properties for medicinal and agrochemical applications.
Chien‐Chi Hsiao, Hsuan‐Hung Liao, Magnus Rueping
Angew. Chem. Int. Ed. 2014, 53, 13258-13263

ABSTRACT:
A protocol for the highly enantioselective synthesis of 9-substituted tetrahydroxanthenones by means of asymmetric Brønsted acid catalysis has been developed. A chiral binol-based N-triflyphosphoramide was found to promote the in situ generation of ortho-quinone methides and their subsequent reaction with 1,3-cyclohexanedione to provide the desired products with excellent enantioselectivities. In addition, a highly enantio- and diastereoselective Brønsted acid catalyzed desymmetrization of 5-monosubstituted 1,3-dicarbonyl substrates with ortho-quinone methides gives rise to valuable tetrahydroxanthenes containing two distant stereocenters.
Jochen Zoller, David C. Fabry, Meria A. Ronge, Magnus Rueping
Angew. Chem. Int. Ed. 2014, 53, 13264-13268

ABSTRACT:
We report a new combined photoredox- and palladium-catalyzed system for the synthesis of highly functionalized indoles. Using catalytic amounts of a photoredox catalyst in the presence of visible light, the typical high loadings of external oxidants could be avoided. Mechanistic studies revealed for the first time that either a) the photoredox catalyst in the absence of oxygen or b) the in situ formed superoxide anions in the presence of oxygen and photoredox catalyst function as the external oxidant. Notably, the safe and practical method described herein represents an alternative19 to common procedures involving peroxo species. Since only small amounts of the oxidant are generated that are furthermore immediately consumed, side reactions of substrate or product can be avoided. Thus, oxidant-sensitive substrates can be used and this makes this method highly suitable for syntheses.
Matthias Leiendecker, Chien‐Chi Hsiao, Lin Guo, Nurtalya Alandini, Magnus Rueping
Angew. Chem. Int. Ed. 2014, 53, 12912-12915

ABSTRACT:
The direct replacement of aromatic methoxy groups with activated carbon nucleophiles would give rise to novel synthetic pathways for targeted and diversity-oriented syntheses. We demonstrate here that this transformation can be achieved in a one-step reaction involving a bifunctional organolithium nucleophile in combination with a C-OMe bond-cleaving nickel catalyst. The resulting products are stable, α-CH active, and suitable for various further modifications. The new method allows direct replacement of aromatic methoxy groups with bifunctional nucleophiles. Synthetically, this can be achieved in a one-step nickel-catalyzed reaction of aryl ethers, whereby the products are obtained in good yields under mild reaction conditions. The products formed are stable, α-C active, and suitable for various further modifications. To demonstrate the utility of the developed method, several Ar-OMe ethers were directly transformed into diverse valuable synthetic building blocks in good yields.
Uxue Uria, Carlos Vila, Ming‐Yuan Lin, Magnus Rueping
Chem. Eur. J. 2014, 20, 13913-13917

ABSTRACT:
The enantioselective synthesis of α- and γ-tocopherol (the most biologically active members of vitamin E family) and analogues has been accomplished employing a new enantioselective gold catalyzed intramolecular allylic alkylation reaction followed by an olefin cross-metathesis as key steps. The methodology proved to be applicable to different olefins highlighting its potential for the synthesis of diverse libraries. We have developed a new synthesis of α- and γ-tocopherol as well as analogues employing a novel asymmetric gold-catalyzed intramolecular allylic alkylation reaction and an olefin cross-metathesis as key steps. The methodology presented herein proved to be compatible with different olefins, highlighting its potential for the synthesis of diverse libraries of valuable biologically active compounds. Furthermore, the new gold-catalyzed intramolecular asymmetric allylic alkylation provides various chromans with quaternary stereocenters with excellent yields and high levels of enantioselectivity.
Xin Hong, Hatice Başpınar Küçük, Modhu Sudan Maji, Yun-Fang Yang, Magnus Rueping, K. N. Houk
J. Am. Chem. Soc. 2014, 136, 13769-13780

ABSTRACT:
Brønsted acid catalyzed (3+ + 2) cycloadditions between hydrazones and alkenes provide a general approach to pyrazolidines. The acidity of the Brønsted acid is crucial for the catalytic efficiency: the less acidic phosphoric acids are ineffective, while highly acidic chiral N-triflylphosphoramides are very efficient and can promote highly enantioselective cycloadditions. The mechanism and origins of catalytic efficiencies and selectivities of these reactions have been explored with density functional theory (M06-2X) calculations. Protonation of hydrazones by N-triflylphosphoramide produces hydrazonium–phosphoramide anion complexes. These ion-pair complexes are very reactive in (3+ + 2) cycloadditions with alkenes, producing pyrazolidine products. Alternative 1,3-dipolar (3 + 2) cycloadditions with the analogous azomethine imines are much less favorable due to the endergonic isomerization of hydrazone to azomethine imine. With N-triflylphosphoramide catalyst, only a small distortion of the ion-pair complex is required to achieve its geometry in the (3+ + 2) cycloaddition transition state. In contrast, the weak phosphoric acid does not protonate the hydrazone, and only a hydrogen-bonded complex is formed. Larger distortion energy is required for the hydrogen-bonded complex to achieve the “ion-pair” geometry in the cycloaddition transition state, and a significant barrier is found. On the basis of this mechanism, we have explained the origins of enantioselectivities when a chiral N-triflylphosphoramide catalyst is employed. We also report the experimental studies that extend the substrate scope of alkenes to ethyl vinyl ethers and thioethers.
Dixit Parmar, Erli Sugiono, Sadiya Raja, Magnus Rueping
Chem. Rev. 2014, 114, 9047-9153

ABSTRACT:
This review focuses on the recent progress of the methodologies utilizing BINOL-derived Brønsted acids. Brønsted acid catalysis is one of the wide-ranging disciplines to exist in organic chemistry. The asymmetric division within this field has received a tremendous amount of attention from research groups all around the world, and now it can truly be considered as an essential tool for any organic chemist. Currently, the choice of chiral Brønsted acids in the literature isvast and varied; however, one of the most powerful andprominent class of catalysts that are utilized are the BINOL-derived phosphoric acids and their related family members. Chiral BINOL-derived Brønsted acids have shown them-selves to be highly efficient catalysts for a huge plethora of transformations and allow the end user to form C−C, C−H, and a variety of C−X bonds in a highly enantioselective fashion. lthough within this category phosphoric acids arestrongly known for activating imine substrates, stronger acids inthe form ofN-triflyl phosphoramides have bridged the gapsomewhat to accessing previously thought out-of-reachsubstrates. Their utility in synthesis however is not solelylimited to their acidic character, and more recently they havebecome extremely powerful chiral counterions for an increasinglist of reactions. Furthermore, they can be combined with metalcatalysts to create a synergistic effect, which has opened newreaction modes previously not possible with the individualcatalysts themselves. mproved understanding of the mechanisms and inter-actions associated between the catalyst and the substrates hasallowed research groups to develop highly powerful method-ologies. Unfortunately, our understanding is still far fromcomplete, and currently we have a crude understanding ofhow the catalysts function, but detailed experimental andcomputational studies are still required for further progress inthefield. Our knowledge on the exact nature ofenantioselectivity is also lacking, and hence for a largeselection of reactions we are unable to predict the absolutestereochemistry prior to measurement. On a critical note, it isstill common tofind methodologies that utilize high catalystloadings, and this may be a parameter that can be improvedwith better understanding. Having said that, since theirintroduction as organocatalysts, research groups haveprovided a rich wealth of methodologies that can be catalyzedby BINOL-derived Brønsted acids. They have covered a widerange of topics and have continued to push the boundaries ofwhat can be achieved. We anticipate that the ability and scopeof these catalysts will grow in the future, and we envision thatthe following decade will be equally exciting for the synthetic organic community.
David C. Fabry, Jochen Zoller, Sadiya Raja, Magnus Rueping
Angew. Chem. Int. Ed. 2014, 53, 10228-10231

Direct, oxidative metal-catalyzed C-H functionalizations of arenes are important in synthetic organic chemistry. Often, (over-)stoichoimetric amounts of organic or inorganic oxidants have to be used in these reactions. The combination of rhodium and photoredox catalysis with visible light allows the direct C-H olefination of arenes. Small amounts (1 mol %) of a photoredox catalyst resulted in the efficient C-H functionalization of a broad range of substrates under mild conditions. The dual catalysis concept for the ortho olefination of aryl amides and its applicability to a broad range of substrates was highlighted. The previously often used (over)stoichiometric amounts of oxidants can be readily substituted with a photoredox catalyst, which makes this system both attractive and a more environmentally sound alternative. Further attempts to apply this new type of combined catalysis to other substrate classes and metal catalysts are currently underway.
Pavlo Nikolaienko, Roman Pluta, Magnus Rueping
Chem. Eur. J. 2014, 20, 9867-9870

ABSTRACT:
A direct process for the trifluoromethylthiolation of allylic and benzylic alcohols under mild conditions has been developed. A wide range of free alcohols underwent nucleophilic substitution in the presence of stable CuSCF3 and BF3⋅Et2O to give the corresponding products in good to excellent yields. The new method for the synthesis of trifluoromethyl thioethers starting from readily available allylic and benzylic alcohols. In contrast to earlier reports, no prefunctionalization of the alcohols is necessary. A wide variety of differently substituted substrates were applied in the reaction with CuSCF3 to give the corresponding products in good to high yields, within short reaction times. The developed reaction procedure shows broad scope, uses safe and nontoxic metals and reagents, and can be carried out under mild reaction conditions, making it a useful reaction for preparative organic synthesis.
Hong Hou, Shaoqun Zhu, Fangfang Pan, Magnus Rueping
Org. Lett. 2014, 16, 2872-2875

ABSTRACT:
A new oxidative [3 + 2] cycloaddition of N-substituted hydroxylamines with alkenes was established under visible light photoredox catalysis. This novel protocol provides a rapid, mild, and efficient access to valuable five-membered ring isoxazolidine heterocycles in a concise fashion. We have developed for the first time a visible-light mediated oxidative nitrone formation/[3 + 2] cycloaddition reaction for the preparation of isoxazolidines. This novel protocol provides a rapid, mild, and efficient access to important five-membered ring isoxazolidine heterocycles in a concise fashion. Notably, the aerobic oxidation of hydroxylamines to nitrones can be achieved through visible light photoredox catalysis under environmentally benign conditions, with low catalyst loading (1 mol %) and no additional oxidant. The application of this powerful system to the synthesis of natural products and pharmaceuticals is currently underway in our laboratories.
12. Introduction to ACS Catalysis Virtual Special Issue on Cascade Catalysis
Wolfgang Kroutil, Magnus Rueping
ACS Catal. 2014, 4, 2086-2087

ABSTRACT:
This virtual special issue of ACS Catalysis on Cascade Catalysis includes studies that cover different recent approaches in cascade catalysis, including established and emerging methodologies.
Carlos Vila, Jonathan Lau, Magnus Rueping
Beilstein J. Org. Chem. 2014, 10, 1233–1238

ABSTRACT:
Pyrrolo[2,1-a]isoquinoline alkaloids have been prepared via a visible light photoredox catalyzed oxidation/[3 + 2] cycloaddition/oxidative aromatization cascade using Rose Bengal as an organo-photocatalyst. A variety of pyrroloisoquinolines have been obtained in good yields under mild and metal-free reaction conditions. We have developed a metal-free photoredox oxidation/[3 + 2] dipolar cycloaddition/oxidative aromatization cascade catalyzed by Rose Bengal using visible-light. This protocol offers a “green” and straightforward synthesis of pyrrolo[2,1-a]isoquinolines starting from readily available maleimides and tetrahydroisoquinolines.
David C. Fabry, Erli Sugiono, Magnus Rueping
Isr. J. Chem. 2014, 54, 341-350

ABSTRACT:
This review provides an overview of recent developments in the area of continuous flow process optimization by employing self-optimizing reactor systems. Although only few reactor concepts have been realized to date, impressive progress has been made, which now allows fully automated reaction optimization without the need of human interactions. The integration of feedback algorithms into continuous flow reactors allows the control of different reaction parameters at the same time, resulting in self-optimizing processes that are completely independent from human interactions. As a result, savings in cost and time that have not previously been possible in such a manner can be achieved. By establishing such systems, a big step towards the automated syntheses of bulk or fine chemicals has been made. Having shown several examples of different algorithms, each one chosen for the specific investigation, self-optimization could be accomplished for rather complex, multi-step syntheses, including those of chiral compounds, as well as in larger scale synthesis. The combination of the appropriate analytic tool and a suitable microreactor setup proved crucial in guaranteeing a perfect interplay of all components, resulting in rapid autonomous reaction optimization.
Magnus Rueping, Jeremy Dufour, Lan Bui
ACS Catal. 2014, 4, 1021-1025

ABSTRACT:
A convergent catalysis approach has been developed. The combined metal-catalyzed and organocatalyzed cascade consists of two oxidations, an aza-Michael addition and an aldol condensation, and involves a multicatalysis approach to provide 1,2-dihydroquinolines in a highly enantioselective fashion. We have developed a new convergent catalysis approach by combining two catalytic oxidative cycles in one pot with iminium/enamine catalysis. The convergence of this process is caused by the oxidation of two substrates, an allylic alcohol and a 2-amino benzyl alcohol, by the substrate-selective redox TPAP/NMO system. This catalytic oxidation, compatible with diarylprolinolsilyl ether catalysis, allows the in situ generation of two aldehydes that react subsequently in an enantioselective Michael addition/intramolecular aldol/dehydration pathway. The domino reaction proved to be very efficient and general, affording a wide range of enantiopure N-protected 1,2-dihydroquinolines under mild and operationally simple conditions. This original process widens the scope of secondary amine catalysis as alcohols can now be used as starting materials instead of more sensitive aldehydes. Furthermore, our oxidative iminium/enamine cascade enhances the versatility of asymmetric covalent catalysis by exploiting the compatibility of metal catalysis with organocatalysis. Finally, the demonstrated convergent catalysis concept will be a blue print for many possible reaction combinations involving one-pot multicomponent multicatalysis.
Chandra M. R. Volla, Iuliana Atodiresei, Magnus Rueping
Chem. Rev. 2014, 114, 2390-2431

ABSTRACT:
The review highlighted the different advantages associated with organocatalytic cascade reactions and presented the classification of various cascade reactions useful for carbon–carbon bond formation, based on the number of transformations.
Roman Pluta, Pavlo Nikolaienko, Magnus Rueping
Angew. Chem. Int. Ed. 2014, 53, 1650-1653

ABSTRACT:
A new and safe method for the synthesis of N-(trifluoromethylthio)phthalimide, a convenient and shelf-stable reagent for the direct trifluoromethylthiolation, has been developed. N-(Trifluoromethylthio)phthalimide can be used as an electrophilic source of F3CS+ and reacts readily with boronic acids and alkynes under copper catalysis. The utility of CF3S-containing molecules as biologically active agents, the mild reaction conditions employed, and the high tolerance of functional groups demonstrate the potential of this new methodology to be widely applied in organic synthesis as well as industrial pharmaceutical and agrochemical research and development. We have developed for the first time a new and safe method for the synthesis of N-(trifluoromethylthio)phthalimide, a convenient and shelf-stable reagent for the direct trifluoromethylthiolation of readily available boronic acids and alkynes. Because of the utility of the CF3S group in biologically active agents, the mild reaction conditions employed, and high tolerance of functional groups, the method developed has the potential of being widely applied in organic synthesis as well as industrial pharmaceutical and agrochemical research and development.
6. Catalytic and Asymmetric Fluorolactonisations of Carboxylic Acids through Anion Phase Transfer
Dixit Parmar, Modhu Sudan Maji, Magnus Rueping
Chem. Eur. J. 2014, 20, 83-86

ABSTRACT:
Catalytic fluorolactonisations of aromatic carboxylic acids have been developed. The reactions proceed under mild conditions using the commercially available reagent Selectfluor. A weak phase transfer of the reagent mediated by Na2CO3 allows the reaction to be conducted in non-polar solvents. Furthermore, by the use of a catalytic amount of (DHQ)2PHAL (hydroquinine 1,4-phthalazinediyl diether), the first asymmetric fluorolactonisation has been achieved. The corresponding isobenzofuran core can be found in many biologically active molecules. We have developed the first fluorolactonisations of aromatic carboxylic acids to give fluorinated isobenzofurans. Mechanistic studies suggest that the reaction proceeds through a weak phase transfer, which allows non-polar solvents to be used and allows the products to be formed in high yields. Furthermore, preliminary experiments have demonstrated that in combination with a cinchona alkaloid the reaction can be carried out in an asymmetric manner for the first time. The reaction uses a mild and safe reagent with an operationally simple procedure. The scope, mechanism and asymmetric transformation are currently being investigated in our laboratory and will be reported in due course.
5. Mild and metal-free oxy- and amino-fluorination for the synthesis of fluorinated heterocycles
Dixit Parmara, Magnus Rueping
Chem. Commun. 2014, 50, 13928-13931

ABSTRACT:
A mild intramolecular fluoro-cyclisation reaction of benzylic alcohols and amines has been developed. This strategy uses commercially available Selectfluor to trigger electrophilic cyclisations to afford fluorinated heterocycles containing 1,3-disubstitution. The dual role of the reagent as a fluorine source and a base is shown to be crucial for reactivity. We have developed an operationally simple and mild route to access fluorinated isobenzofurans and isoindolines. The reaction involves an electrophilic oxy- and amino-fluorination process mediated by commercially available Selectfluor. The protocol has been shown to be useful for accessing 1,3-substituted products containing both alkyl and aromatic substituents. In addition to being metal-free, the methodology requires no additional base by exploiting the basic by-product of the reagent Selectfluor as a proton scavenger. The scope, utility and asymmetric transformation is currently being investigated in our laboratory and will be reported in due course.
Chandra M. R. Volla, Arindam Das, Iuliana Atodireseia, Magnus Rueping
Chem. Commun. 2014, 50, 7889-7892

ABSTRACT:
An efficient Brønsted acid assisted Lewis base catalysis protocol for the synthesis of enantiomerically pure trifluoromethylated dihydropyridazines starting from readily available hydrazones and α,β-unsaturated aldehydes has been developed. The reaction exhibits high tolerance towards many functional groups and is applicable to various aliphatic, aromatic and hetero-aromatic α,β-unsaturated aldehydes, and provides the products in good yields and with excellent enantioselectivities.
3. Visible light photoredox-catalysed intermolecular radical addition of α-halo amides to olefins
Masaki Nakajima, Quentin Lefebvrea, Magnus Rueping
Chem. Commun. 2014, 50, 3619-3622

ABSTRACT:
We present α-chloro amides as a new class of α-acetyl radical precursors, which undergo a tin-free, photoredox-catalysed intermolecular α-alkylation with various olefins exclusively in an anti-Markovnikov fashion. The reaction represents a reductive atom transfer radical addition (ATRA) and provides a series of alkylated amides in good yields. W e have developed a protocol for tin-free alkylation of α-chloro amides utilizing photoredox catalysis in combination with visible light. Interestingly, depending on the catalyst used, clean dehalogenation or effective alkylation can be achieved. The alkylation is compatible with a broad spectrum of amides and olefins. The corresponding products are obtained in good to excellent yields with exclusive anti-Markovnikov selectivity
Magnus Rueping, Xiangqian Liu, Teerawut Bootwicha, Roman Pluta, Carina Merkens
Chem. Commun. 2014, 50, 2508-2511

ABSTRACT:
The organocatalytic enantioselective trifluoromethylthiolation of oxindoles employing N-(trifluoromethylthio)phthalimide as a stable, easy to handle CF3S-source has been developed. Optically active products with a quaternary stereogenic center bearing a CF3S-group are obtained in good yields and with good to excellent enantioselectivities. We have developed a novel asymmetric trifluoromethylthiolation of oxindoles catalyzed by cinchona alkaloids. This transformation utilized air and moisture stable N-(trifluoromethylthio)phthalimide as the SCF3 source. A series of optically active oxindoles bearing a chiral SCF3-substituted quaternary stereogenic center were obtained in good yields and with good to excellent enantioselectivities.
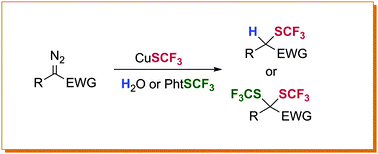
1. Hydrotrifluoromethylthiolation of α-diazo esters – synthesis of α-SCF3 substituted esters
Quentin Lefebvre, Eleonora Fava, Pavlo Nikolaienko, Magnus Rueping
Chem. Commun. 2014, 50, 6617-6619
ABSTRACT:
A practical protocol for hydrotrifluoromethylthiolation of diazo compounds has been developed. A range of diazo compounds in combination with a nucleophilic SCF3 source provided access to valuable trifluoromethylthiolated compounds. Furthermore, a methodology for the first double trifluoromethylthiolation was developed. We have developed a convenient method for the hydrotrifluoromethylthiolation of various diazo compounds. The reaction tolerates most of the common functional groups, and the use of readily available and easy to handle CuSCF3 under mild conditions ensures potential for applications in post-functionalization of complex drug-like molecules. Furthermore, we were able to expand the methodology to the first double trifluoromethylthiolation providing novel fluorinated products in good yield.