
2020
32. Solution processable metal–organic frameworks for mixed matrix membranes using porous liquids
Alexander Knebel, Anastasiya Bavykina, Shuvo Jit Datta, Lion Sundermann, Luis Garzon-Tovar, Yury Lebedev, Sara Durini, Rafia Ahmad, Sergey M Kozlov, Genrikh Shterk, Madhavan Karunakaran, Ionela Daniela Carja, Dino Simic, Irina Weilert, Manfred Klüppel, Ulrich Giese, Luigi Cavallo, Magnus Rueping, Mohamed Eddaoudi, Jürgen Caro, Jorge Gascon
Nat. Mater. 2020, 19, 1346–1353

ABSTRACT:
The combination of well-defined molecular cavities and chemical functionality makes crystalline porous solids attractive for a great number of technological applications, from catalysis to gas separation. However, in contrast to other widely applied synthetic solids such as polymers, the lack of processability of crystalline extended solids hampers their application. In this work, we demonstrate that metal–organic frameworks, a type of highly crystalline porous solid, can be made solution processable via outer surface functionalization using N-heterocyclic carbene ligands. Selective outer surface functionalization of relatively large nanoparticles (250 nm) of the well-known zeolitic imidazolate framework ZIF-67 allows for the stabilization of processable dispersions exhibiting permanent porosity. The resulting type III porous liquids can either be directly deployed as liquid adsorbents or be co-processed with state-of-the-art polymers to yield highly loaded mixed matrix membranes with excellent mechanical properties and an outstanding performance in the challenging separation of propylene from propane. We anticipate that this approach can be extended to other metal–organic frameworks and other applications.
31. Understanding high-salt and cold adaptation of a polyextremophilic enzyme
Ram Karan, Sam Mathew, Reyhan Muhammad, Didier B Bautista, Malvina Vogler, Jorg Eppinger, Romina Oliva, Luigi Cavallo, Stefan T Arold, Magnus Rueping
Microorganisms 2020, 8, 1594

ABSTRACT:
The haloarchaeon Halorubrum lacusprofundi is among the few polyextremophilic organisms capable of surviving in one of the most extreme aquatic environments on Earth, the Deep Lake of Antarctica (−18 °C to +11.5 °C and 21–28%, w/v salt content). Hence, H. lacusprofundi has been proposed as a model for biotechnology and astrobiology to investigate potential life beyond Earth. To understand the mechanisms that allow proteins to adapt to both salinity and cold, we structurally (including X-ray crystallography and molecular dynamics simulations) and functionally characterized the β-galactosidase from H. lacusprofundi (hla_bga). Recombinant hla_bga (produced in Haloferax volcanii) revealed exceptional stability, tolerating up to 4 M NaCl and up to 20% (v/v) of organic solvents. Despite being cold-adapted, hla_bga was also stable up to 60 °C. Structural analysis showed that hla_bga combined increased surface acidity (associated with halophily) with increased structural flexibility, fine-tuned on a residue level, for sustaining activity at low temperatures. The resulting blend enhanced structural flexibility at low temperatures but also limited protein movements at higher temperatures relative to mesophilic homologs. Collectively, these observations help in understanding the molecular basis of a dual psychrophilic and halophilic adaptation and suggest that such enzymes may be intrinsically stable and functional over an exceptionally large temperature range.
Bholanath Maity, Chen Zhu, Huifeng Yue, Long Huang, Moussab Harb, Yury Minenkov, Magnus Rueping, Luigi Cavallo
J. Am. Chem. Soc. 2020, 142, 16942–16952

ABSTRACT:
We report here a comprehensive computational analysis of the mechanisms of the photoredox-nickel-HAT (HAT: hydrogen atom transfer) catalyzed arylation and alkylation of α-amino Csp3–H bonds developed by MacMillan and co-workers. Different alternatives for the three catalytic cycles were tested to identify unambiguously the operative reaction mechanism. Our analysis indicated that the IrIII photoredox catalyst, upon irradiation with visible light, can be either reduced or oxidized by the HAT and nickel catalysts, respectively, indicating that both reductive and oxidative quenching catalytic cycles can be operative, although the reductive cycle is favored. Our analysis of the HAT cycle indicated that activation of a α-amino Csp3–H bond of the substrate is facile and selective relative to activation of a β-amino Csp3–H bond. Finally, our analysis of the nickel cycle indicated that both arylation and alkylation of α-amino Csp3–H bonds occurs via the sequence of nickel oxidation states NiI–NiII–NiI–NiIII and of elementary steps: radical addition-SET-oxidative addition-reductive elimination.
Jan Sklyaruk, Viktoriia Zubar, Jannik C Borghs, Magnus Rueping
Org. Lett. 2020, 22, 6067–6071

ABSTRACT:
The first base metal-catalyzed transfer hydrogenation of alkynes with methanol is described. An air and moisture stable manganese pincer complex catalyzes the reduction of a variety of different alkynes to the corresponding (Z)-olefins in high yields. The reaction is stereo- and chemoselective and scalable.
28. Regiodivergent hydroborative ring opening of epoxides via selective C–O bond activation
Marc Magre, Eva Paffenholz, Bholanath Maity, Luigi Cavallo, Magnus Rueping
J. Am. Chem. Soc. 2020, 142, 14286–14294

ABSTRACT:
A magnesium-catalyzed regiodivergent C–O bond cleavage protocol is presented. Readily available magnesium catalysts achieve the selective hydroboration of a wide range of epoxides and oxetanes yielding secondary and tertiary alcohols in excellent yields and regioselectivities. Experimental mechanistic investigations and DFT calculations provide insight into the unexpected regiodivergence and explain the different mechanisms of the C–O bond activation and product formation.
27. Chemoselective Hydrogenation of Alkynes to (Z)-Alkenes Using an Air-Stable Base Metal Catalyst
Viktoriia Zubar, Jan Sklyaruk, Aleksandra Brzozowska, Magnus Rueping
Org. Lett. 2020, 22, 5423–5428

ABSTRACT:
A highly selective hydrogenation of alkynes using an air-stable and readily available manganese catalyst has been achieved. The reaction proceeds under mild reaction conditions and tolerates various functional groups, resulting in (Z)-alkenes and allylic alcohols in high yields. Mechanistic experiments suggest that the reaction proceeds via a bifunctional activation involving metal–ligand cooperativity.
Valmik S Shinde, Manoj V Mane, Luigi Cavallo, Magnus Rueping
Chem. Eur. J. 2020, 26, 8308-8313
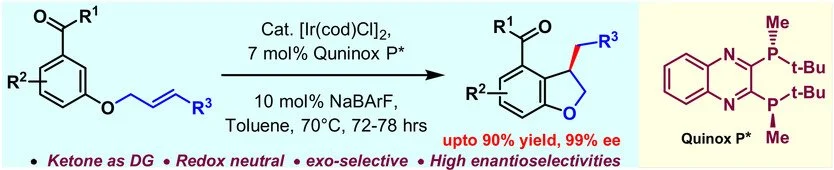
ABSTRACT:
A new catalytic enantioselective hydroarylation of unactivated olefins is developed that provides rapid access to functionalized chiral dihydrobenzofurans with good yields and excellent enantioselectivities. Simple aromatic ketones or amides act as a directing group allowing the regioselective reaction at the more hindered ortho position. Tertiary benzylic stereocenters are obtained directly under mild reaction conditions and with complete atom economy from readily available starting materials.
25. Tuning optical properties of BODIPY dyes by pyrrole conjugation for photoacoustic imaging
Jean Michél Merkes, Twan Lammers, Rajesh Kancherla, Magnus Rueping, Fabian Kiessling, Srinivas Banala
Adv. Optical Mater. 2020, 8, 1902115

ABSTRACT:
Photoacoustic imaging (PAI) is increasingly employed in (pre-) clinical research, thus, development of suitable contrast agents, in particular fluorescence-quenched chromophores, for PAI is of high importance. Small molecule dyes are appropriate due to favorable circulation, excretion properties, and ease of conjugation to targeting moieties. The BODIPY chromophores have been widely used in bioimaging, yet they are not ideal for PAI due to high fluorescence. Hence, here nonfluorescent BODIPY are designed by 1H-pyrrole conjugation (PyBODIPY) to apply as probes for PAI. The PyBODIPYs exhibit absorption maxima up to 800 nm, and PA signal could be detected in concentrations of 1 nmol mL−1 and 35 pmol mm−3, by tube and tissue phantom, respectively. In addition to nonfluorescent, PyBODIPYs are non-phototoxic, photostable, and show high molar extinction coefficients, as well as inertness toward nucleophilic addition. PyBODIPYs are modified with PEG-400, to improve aqueous solubility and to enable in vivo imaging. Thus, PyBODIPY is an attractive small molecule to use as PA contrast agent, which could be coupled to targeting ligands for in vivo use. In addition, 1H-pyrrole conjugation might be applied to the design of novel near-infrared ranged quenchers suitable for PAI, and promote the development of probes for clinical translation.
Jannik C Borghs, Viktoriia Zubar, Luis Miguel Azofra, Jan Sklyaruk, Magnus Rueping
Org. Lett. 2020, 22, 4222–4227

ABSTRACT:
The first base metal-catalyzed regioselective dehydrogenative alkylation of indolines using readily available alcohols as the alkylating reagent is reported. A single air- and moisture-stable manganese catalyst provides access to either C3- or N-alkylated indoles depending on the solvent used. Mechanistic studies indicate that the reaction takes place through a combined acceptorless dehydrogenation and hydrogen autotransfer strategy.
Viktoriia Zubar, Jannik C Borghs, Magnus Rueping
Org. Lett. 2020, 22, 3974–3978

aBSTRACT:
A highly chemoselective base-metal catalyzed hydrogenation and acceptorless dehydrogenation of N-heterocycles is presented. A well-defined Mn complex operates at low catalyst loading (as low as 2 mol %) and under mild reaction conditions. The described catalytic system tolerates various functional groups, and the corresponding reduced heterocycles can be obtained in high yields. Experimental studies indicate a metal-ligand cooperative catalysis mechanism.
22. CoVID-19: Where we are, what we should do and what we should learn
Jörg Eppinger, Magnus Rueping
Preprints 2020, 2020040484 (doi: 10.20944/preprints202004.0484.v1)

aBSTRACT:
There were warnings before; nevertheless the current CoVID-19 pandemic took the world by surprise: within just four month, it conquered the globe and claimed over 200'000 lives. Unprecedented governmental actions put about half of the population under curfew or lock-down. The resulting economic meltdown is expected to eliminate globally 9’000’000’000’000 (9 trillion) USD in 2020 and 2021 alone, a value roughly the size of the yearly GDP of the world’s 150 smallest economies. The resulting crises might cause mass-unemployment and a hunger pandemic later this year. This Essay analyses current statistical data of the CoVID-19 pandemic to develop a guideline for a path through the crisis, minimizing both loss of lives and economic costs. Part 1 details the current situation; part 2 develops a small set of measures, allowing a near normal life until a future vaccination campaign has reached sufficient numbers of people; and part 3 provides some important lessons for the future beyond SARS-CoV-2. The Essay leads to the following key-messages: 1) The CoVID-19 pandemic will stay for at least two more years. This is the minimum time required for a vaccination campaign to reach sufficient numbers of people. 2) The crucial element to control the pandemic is keeping case numbers under the threshold required for a functional tracing, testing & isolation (TTI) strategy. That threshold differs from country to country and strongly depends on culture and the applied tracing technology as well as available testing capacities. 3) The economic burden of a TTI strategy is moderate while fatalities are also reduced. Hence, such an approach is strongly recommended. Its implementation requires a set of simple and cost-effective measures (see figure below), which in combination seem to be sufficient to keep CoVID-2’s reproductive rate at or below 1. 4) Implementing international coordination of actions will be necessary for effective infection-chain tracing5) If case numbers are above the TTI threshold, shutdown measures remain the only option until tracing of infection chains becomes feasible again.6) In the future, neglected pandemic-related research requires a funding boost. Just 1% of the bill of the current crisis could support the research of 45’000 scientist for 20 years.
21. Crystal structure and active site engineering of a halophilic γ-carbonic anhydrase
Malvina Vogler, Ram Karan, Dominik Renn, Alexandra Vancea, Marie-Theres Vielberg, Stefan W Grötzinger, Priya DasSarma, Shiladitya DasSarma, Jörg Eppinger, Michael Groll, Magnus Rueping
Front. Microbiol. 2020, 11, 742

aBSTRACT:
Environments previously thought to be uninhabitable offer a tremendous wealth of unexplored microorganisms and enzymes. In this paper, we present the discovery and characterization of a novel γ-carbonic anhydrase (γ-CA) from the polyextreme Red Sea brine pool Discovery Deep (2141 m depth, 44.8°C, 26.2% salt) by single-cell genome sequencing. The extensive analysis of the selected gene helps demonstrate the potential of this culture-independent method. The enzyme was expressed in the bioengineered haloarchaeon Halobacterium sp. NRC-1 and characterized by X-ray crystallography and mutagenesis. The 2.6 Å crystal structure of the protein shows a trimeric arrangement. Within the γ-CA, several possible structural determinants responsible for the enzyme’s salt stability could be highlighted. Moreover, the amino acid composition on the protein surface and the intra- and intermolecular interactions within the protein differ significantly from those of its close homologs. To gain further insights into the catalytic residues of the γ-CA enzyme, we created a library of variants around the active site residues and successfully improved the enzyme activity by 17-fold. As several γ-CAs have been reported without measurable activity, this provides further clues as to critical residues. Our study reveals insights into the halophilic γ-CA activity and its unique adaptations. The study of the polyextremophilic carbonic anhydrase provides a basis for outlining insights into strategies for salt adaptation, yielding enzymes with industrially valuable properties, and the underlying mechanisms of protein evolution.
20. Photoacoustic Imaging Probes Based on Tetrapyrroles and Related Compounds
Jean Michel Merkes, Leiming Zhu, Srishti Ballabh Bahukhandi, Magnus Rueping, Fabian Kiessling, Srinivas Banala
Int. J. Mol. Sci. 2020, 21, 3082
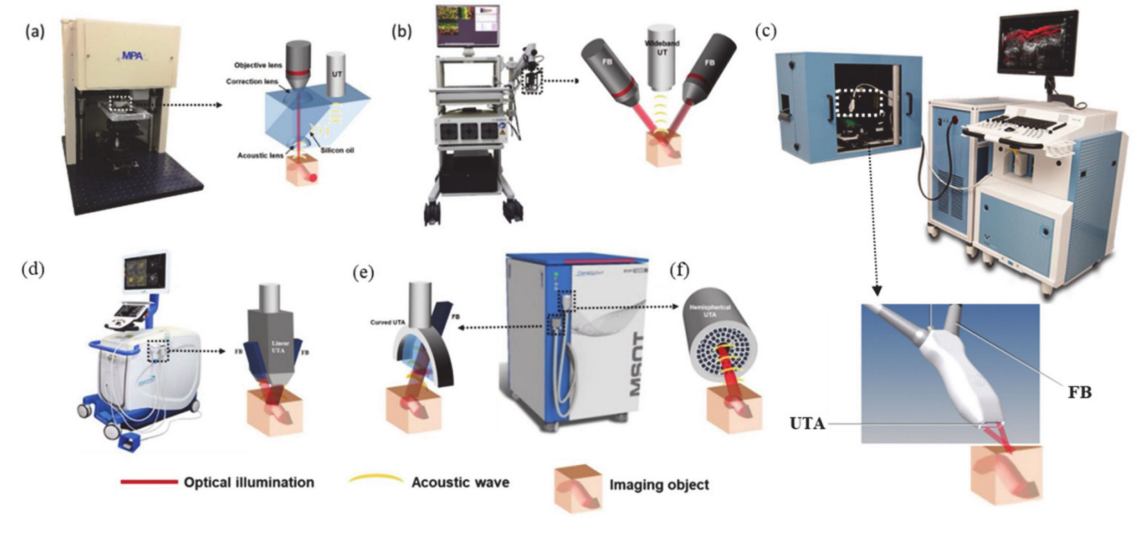
aBSTRACT:
Photoacoustic imaging (PAI) is a rapidly evolving field in molecular imaging that enables imaging in the depths of ultrasound and with the sensitivity of optical modalities. PAI bases on the photoexcitation of a chromophore, which converts the absorbed light into thermal energy, causing an acoustic pressure wave that can be captured with ultrasound transducers, in generating an image. For in vivo imaging, chromophores strongly absorbing in the near-infrared range (NIR > 680 nm) are required. As tetrapyrroles have a long history in biomedical applications, novel tetrapyrroles and inspired mimics have been pursued as potentially suitable contrast agents for PAI. The goal of this review is to summarize the current state of the art in PAI applications using tetrapyrroles and related macrocycles inspired by it, highlighting those compounds exhibiting strong NIR-absorption. Furthermore, we discuss the current developments of other absorbers for in vivo photoacoustic (PA) applications.
19. Chemoselective Hydroboration of Propargylic Alcohols and Amines Using a Manganese (II) Catalyst
Aleksandra Brzozowska, Viktoriia Zubar, Ruth-Christine Ganardi, Magnus Rueping
Org. Lett. 2020, 22, 3765–3769

aBSTRACT:
The first manganese-catalyzed hydroboration of propargylic alcohols and amines as well as internal alkynes is reported. High regio- and stereoselectivity is achieved by applying 2 mol % of a manganese precatalyst based on the readily accessible bis(imino)pyridine ligand and MnCl2 as metal source. Propargylic alcohols and amines, as well as symmetric internal alkynes, were efficiently converted into the corresponding functionalized alkenes, which can serve as important and valuable intermediates for further synthetic applications such as cross-coupling reactions.
Marc Magre, Marcin Szewczyk, Magnus Rueping
Org. Lett. 2020, 22, 3209–3214

aBSTRACT:
A new reduction of carbamates to N-methyl amines is presented. The magnesium-catalyzed reduction reaction allows the conversion of cyclic and linear carbamates, including N-Boc protected amines, into the corresponding N-methyl amines and amino alcohols which are of significant interest due to their presence in many biologically active molecules. Furthermore, the reduction can be extended to the formation of N-trideuteromethyl labeled amines.
17. Remote trifluoromethylthiolation enabled by organophotocatalytic C–C bond cleavage
Tengfei Ji, Xiang-Yu Chen, Long Huang, Magnus Rueping
Org. Lett. 2020, 22, 2579–2583

aBSTRACT:
The first metal-free ring opening/trifluoromethylthiolation of cycloalkanols for the formation of remote C(sp3)–SCF3 bonds has been developed. A variety of trifluoromethylthiolated carbonyl compounds that are otherwise difficult to achieve were prepared in good yields under mild reaction conditions. The reaction is assumed to proceed via C–C bond cleavage of the alkoxyl radical species generated via a photoredox-enabled intramolecular proton-coupled electron transfer process, followed by trifluoromethylthiolation of the resulting C-centered radical with the N-(trifluoromethylthio)phthalimide reagent.
Luis Miguel Azofra, Mai Anh Tran, Viktoriia Zubar, Luigi Cavallo, Magnus Rueping, Osama El-Sepelgy
Chem. Commun., 2020, 56, 9094-9097

aBSTRACT:
An unprecedented base metal catalysed asymmetric synthesis of α-chiral amine precursors from racemic alcohols is reported. This redox-neutral reaction utilises a bench-stable manganese complex and Ellman's sulfinamide as a versatile ammonia surrogate. DFT calculations explain the unusual finding of the highly stereoselective transformation enabled by a catalyst that undergoes an unusual dynamic kinetic resolution.
Jiaqi Jia, Rajesh Kancherla, Magnus Rueping, Long Huang
Chem. Sci., 2020, 11, 4954-4959

aBSTRACT:
A new catalytic method for the direct alkylation of allylic C(sp3)–H bonds from unactivated alkenes via synergistic organo- and photoredox catalysis is described. The transformation achieves an efficient, redox-neutral synthesis of homoallylamines with broad functional group tolerance, under very mild reaction conditions. Mechanistic investigations indicate that the reaction proceeds through the N-centered radical intermediate which is generated by the allylic radical addition to the imine.
Patricia E Krach, Abhishek Dewanji, Tingting Yuan, Magnus Rueping
Chem. Commun., 2020, 56, 6082-6085

aBSTRACT:
Herein, we report a dual catalytic system for the direct benzylic C–H acylation reaction furnishing a variety of unsymmetrical ketones. A benzophenone-derived photosensitizer combined with a nickel catalyst has been established as the catalytic system. Both acid chlorides and anhydrides are able to acylate the benzylic position of toluene and other methylbenzenes. The method offers a valuable alternative to late transition metal catalyzed C–H acylation reactions.A
Chen Zhu, Huifeng Yue, Lingling Chu, Magnus Rueping
Chem. Sci. 2020, 11, 4051-4064
aBSTRACT:
Cascade reactions that produce multiple chemical bonds in one synthetic operation are important in the efficient construction of complex molecules. In addition, photoredox and nickel dual catalysis opens a new and powerful avenue for transition-metal-catalyzed cross-coupling reactions. By combining these two concepts, photoredox and nickel dual-catalyzed cascade reactions have been recently established, and they provide an efficient and mild method for accessing a series of valuable organic compounds.


Chen Zhu, Huifeng Yue, Pavlo Nikolaienko, Magnus Rueping
CCS Chem. 2020, 2, 179–190
aBSTRACT:
We have achieved a nickel-catalyzed cross-coupling reaction via concerted paired electrolysis under mild reaction conditions. In this electrochemical transformation, the anodic oxidation of NiII to NiIII and cathodic reduction of NiI to Ni0 occurred simultaneously, resulting in an economical and sustainable cross-coupling protocol. Moreover, we performed mechanistic investigations, achieved by experiments and density functional theory (DFT) calculations for different C–heteroatom bond formations to reveal the catalytic cycle in more detail.

Sathish Kumar Gadde, Anatoly Peshkov, Aleksandra Brzozowska, Pavlo Nikolaienko, Chen Zhu, Magnus Rueping
Angew. Chem. Int. Ed. 2020, 59, 6513-6519
aBSTRACT:
The first electrochemical approach for nickel-catalyzed cross-electrophile coupling was developed. This method provides a novel route to 1,1-diarylalkane derivatives from simple and readily available alkyl and aryl halides in good yields and excellent regioselectivity under mild conditions. The procedure shows good tolerance for a broad variety of functional groups and both primary and secondary alkyl halides can be used. Furthermore, the reaction was successfully scaled up to the multigram scale, thus indicating potential for industrial application. Mechanistic investigation suggested the formation of a nickel hydride in the electroreductive chain-walking arylation, which led to the development of a new nickel-catalyzed hydroarylation of styrenes to provide a series of 1,1-diaryl alkanes in good yields under mild reaction conditions.
Long Huang, Tengfei Ji, and Magnus Rueping
J. Am. Chem. Soc. 2020, 142, 3532-3539
aBSTRACT:
Cross-coupling reactions for carbon–carbon and carbon–heteroatom bond formation are of great importance in modern chemical synthesis. In addition to classical cross-couplings involving preformed or preactivated coupling partners, more recently breakthroughs have been made in the selective, direct coupling of abundant aliphatic carbon–hydrogen bonds using hydrogen atom transfer reactions in which the bond-dissociation energy is the thermodynamic driving force. The more challenging carbon–carbon bond activation is still rather underdeveloped due to the bond inertness. Herein, we report a mild and general strategy for the activation of a diverse set of readily available cyclic alcohols for the remote and site-specific arylation of ketones via the combination of photoredox-mediated multisite concerted proton–electron transfer (MS-PCET) and nickel catalysis. The current cross-coupling proceeds with the generation of an alkoxy radical utilizing bond-dissociation free energy (BDFE) as the thermodynamic driving force. Subsequently, the resulting remote carbon-centered radicals formed by C–C cleavage merge with the nickel catalytic cycle to create the challenging C(sp3)–C(sp2) bonds.

Abhishek Dewanji, Raoul F. Bülow, and Magnus Rueping
Org. Lett. 2020, 22, 1611-1617
aBSTRACT:
A successful protocol for the reductive aryl–aryl cross-coupling of polyfluorinated arenes with a broad range of aryl halides has been developed. Sequential carbon–fluorine bond cleavage and carbon–carbon bond formation are two of the important features of the reaction. Addition of an aryl radical anion to a nickel intermediate was achieved for the first time using polyfluoroarenes as radical precursors. This, in combination with the excellent para selectivity, paves the way for the synthesis of various new multifluorinated biaryl compounds.

Leiming Zhu, Leonard Himmel, Jean Michél Merkes, Fabian Kiessling, Magnus Rueping, and Srinivas Banala
Chem. Eur. J. 2020, 26, 4232-4235
aBSTRACT:
Atropisomerism has been observed in a variety of biaryl compounds and meso-aryl substituted porphyrins. However, in porphyrins, this phenomenon had been shown only with o-substituted 6-membered aromatic groups at the meso-position. We show herein that a 5-membered heteroaromatic (N-mesyl-pyrrol-2-yl) group at the meso-position leads to atropisomerism. In addition, we report a ‘one-pot’ synthetic route for the synthesis of ‘all-pyrrolic’ porphyrin (APP) with several N-protection groups (Boc, Cbz, Ms and Ts). Among these groups, we found that only the Ms group gave four individually separable atropisomers of meso-tetra(N-Ms-pyrrol-2-yl) porphyrin. Furthermore, the reductive removal of Cbz- was achieved to obtain meso-tetra(pyrrol-2-yl) porphyrin. Thus, our synthetic procedure provides an easy access to a group of APPs and stable atropisomers, which is expected to expand the application of novel APP-based materials.

7. Magnesium-Catalyzed Stereoselective Hydrostannylation of Internal and Terminal Alkynes
Marc Magre, Marcin Szewczyk, and Magnus Rueping
Org. Lett. 2020, 22, 1594-1598
aBSTRACT:
A regio- and stereoselective magnesium-catalyzed hydrostannylation of internal and terminal alkynes has been developed. Excellent yields and selectivities are obtained for a wide range of terminal and internal symmetrical and unsymmetrical alkynes by using this alkaline earth metal catalyst as an effective alternative to transition metal catalysts.

6. Reductive coupling of imines with redox-active esters by visible light photoredox organocatalysis
Jiaqi Jia, Quentin Lefebvre and Magnus Rueping
Org. Chem. Front., 2020, 7, 602-608
aBSTRACT:
The addition of organometallic compounds to imines is a direct way for accessing α-branched secondary amines which are found in numerous bioactive molecules. Although convenient, such reactions typically involve the formation and isolation of the nucleophile in the case of Grignard reactions, or the in situ formation of the nucleophile for Barbier reactions, leading to stoichiometric amounts of metallic wastes. Herein, we report the direct alkylation of imines with redox-active esters by visible light photoorganocatalysis. With Rose bengal as inexpensive photocatalyst, and green light as sustainable energy source, the synthesis of a wide range of amines and θ-amino esters was achieved from inexpensive feedstock chemicals, in a highly modular fashion.

Huifeng Yue, Chen Zhu, Rajesh Kancherla, Fangying Liu, Magnus Rueping
Angew. Chem. Int. Ed. 2020, 59, 5738-5746
aBSTRACT:
Alkynes are an important class of organic molecules due to their utility as versatile building blocks in synthesis. Although efforts have been devoted to the difunctionalization of alkynes, general and practical strategies for the direct hydroalkylation and alkylarylation of terminal alkynes under mild reaction conditions are less explored. Herein, we report a photoredox/nickel dual-catalyzed anti-Markovnikov-type hydroalkylation of terminal alkynes as well as a one-pot arylalkylation of alkynes with alkyl carboxylic acids and aryl bromides via a three-component cross-coupling. The results indicate that the transformations proceed via a new mechanism involving a single-electron transfer with subsequent energy-transfer activation pathways. Moreover, steady-state and time-resolved fluorescence-spectroscopy measurements, density functional theory (DFT) calculations, and wavefunction analysis have been performed to give an insight into the catalytic cycle.

Denise Kleinschmidt, Marta Sofia Fernandes, Matthias Mork, Anna Astrid Meyer, Julian Krischel, Mikhail V. Anakhov, Rustam A. Gumerov, Igor I.Potemkin, Magnus Rueping, Andrij Pich
J. Colloid Interface Sci. 2020, 559, 76-87
aBSTRACT:
Exploring and controlling chemical reactions in compartments opens new platforms for designing bioinspired catalysts and energy-autonomous systems. Aqueous polymer networks or hydrogels serve as a perfect model for biological tissues, allowing systematic investigations of chemical transformations in compartments. Herein, we report the synthesis of a versatile, colloidal microgel catalyst containing covalently bound L-proline as an organocatalyst. The key finding of our work is that the catalytic activity can be tuned by adjusting the distribution of the organocatalyst in the microgel network as well as the properties of the solvent. We demonstrate that L-proline groups integrated into microgels enable the reaction of 4-nitrobenzaldehyde and cyclohexanone in a heterogeneous reaction mixture in which free L-proline is not active. By controlling the localization of the L-proline groups within the microgel network (core or corona), the rate of the aldol reaction in homogenous and heterogeneous reaction mixtures can be modulated. Furthermore, microgels with covalently attached catalysts can be recycled and reused in sequential catalytic runs without deterioration of the catalyst performance in terms of activity and selectivity. The internal structure of the microgel in heterogeneous reaction mixtures was studied by computer simulations.

3. One Amine–3 Tasks: Reductive Coupling of Imines with Olefins in Batch and Flow
Quentin Lefebvre, Riccardo Porta, Anthony Millet, Jiaqi Jia, Magnus Rueping
Chem. Eur. J. 2020, 26, 1363-1367
aBSTRACT:
Owing to their wide range of biological properties, γ-aminobutyric acid derivatives (GABA) have been extensively studied and found noteworthy industrial applications. However, atom-economical and efficient processes for their production are scarce and would greatly benefit from further investigations. Herein, we demonstrate that an iridium-based photocatalyst promotes the direct reductive cross-coupling of imines with olefins upon irradiation with visible light to give GABA derivatives in good yields and selectivities. We also stress the enabling triple role of tributylamine additive in this process, discuss the advantages of strategies based on proton-coupled electron transfer (PCET) and demonstrate the scale-up of this reaction in continuous flow.
2. Hydride Transfer Enables the Nickel‐Catalyzed ipso‐Borylation and Silylation of Aldehydes
Watchara Srimontree, Lin Guo, Magnus Rueping
Chem. Eur. J. 2020, 26, 423-427

aBSTRACT:
Nickel-catalyzed ipso-borylations and silylations of aldehydes are described for the first time. The new functional-group interconversion protocol is characterized by its scalability, functional-group tolerance and wide substrate scope, including examples of late-stage functionalization of complex molecules. The key for the successful reaction outcome is the use of a ketone as a hydride acceptor that intercepts the nickel hydride to undergo a reductive pathway, thus allowing formation of the desired C−B and C−Si bonds.

1. Cascade Cross‐Coupling of Dienes: Photoredox and Nickel Dual Catalysis
Long Huang, Chen Zhu, Liang Yi, Huifeng Yue, Rajesh Kancherla, Magnus Rueping
Angew. Chem. Int. Ed. 2020, 59, 457-464
aBSTRACT:
Chemical transformations based on cascade reactions have the potential to simplify the preparation of diverse and architecturally complex molecules dramatically. Herein, we disclose an unprecedented and efficient method for the cross-coupling of radical precursors, dienes, and electrophilic coupling partners via a photoredox- and nickel-enabled cascade cross-coupling process. The cascade reaction furnishes a diverse array of saturated carbo- and heterocyclic scaffolds, thus providing access to a quick gain in C−C bond saturation.