
Rapid and Scalable Metal Catalyzed meta-C–H Alkylation Enabled by Resonant Acoustic Mixing (RAM)
Arnab Dey, Rajesh Kancherla, Kuntal Pal, Nathan Kloszewski, Magnus Rueping
Commun. Chem. 2024, 7, 295
DOI:10.1038/s42004-024-01390-1
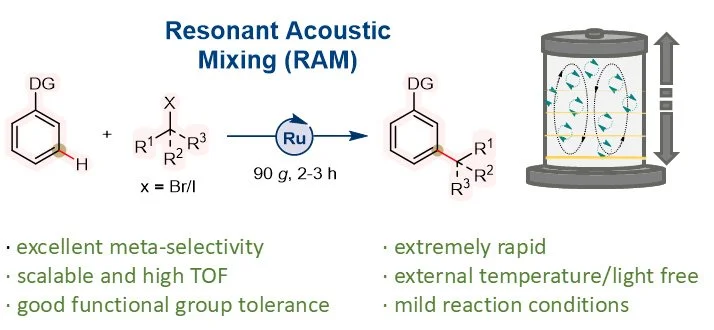
Abstract:
Synthetic chemistry approaches for direct C–H bond alkylation offer a promising alternative to traditional functional-group-centered strategies which often involve multi-step procedures and may suffer from a variety of challenges including scalability. Here, we introduce resonant mixing as an efficient method for meta-C–H alkylation of arenes using a Ru-catalyst, avoiding the need for bulk solvents, external temperature, or light. The described methodology is highly rapid, enabling multigram-scale synthesis of meta-alkylation products within a short reaction time and achieving a very high turnover frequency. For instance, the use of Ru-catalyst (0.5 mol%), ligand (1.0 mol%), and KOAc (5 mol%) produced the alkylated product with a TOF of 146 h-1 at a multigram scale highlighting the efficiency of the RAM in selective C-H functionalization. The reaction operates via a radical mechanism and is characterized by its mild reaction conditions, substrate compatibility, and exceptional meta-selectivity, all while significantly reducing reaction times.
Ru-OV Site-Mediated Product Selectivity Switch for Overall Photocatalytic CO2 Reduction
C. Feng, M. Hu, S. Zuo, J. Luo, P. Castaño, Y. Ren, M. Rueping, H. Zhang
Adv. Mater. 2024, 2411813
DOI:10.1002/adma.202411813

Product Selectivity Switch for Overall Photocatalytic CO2 Reduction
Abstract:
The photocatalytic reduction of carbon dioxide (CO2) to methane (CH4) represents a sustainable route for directly converting greenhouse gases into chemicals but poses a significant challenge in achieving high selectivity due to thermodynamic and kinetic limitations during the reaction process. This work establishes Ru-OV active sites on the surface of TiO2 by anchoring coordination unsaturated Ru single-atoms, which stabilize crucial reaction intermediates and facilitate local mass transfer to achieve dual optimization of the thermodynamics and kinetics of the overall photocatalytic CO2 reduction. Combining operando spectroscopy with density functional theory (DFT) calculations indicates that oxygen vacancies (OV) inhibits the desorption of *CO, whereas Ru facilitates proton extraction. This configuration not only lowers the overall activation energy barrier but has also been engineered to serve as a selectivity switch, changing the reaction route to produce CH4 instead of CO. Consequently, the Ru-OV/TiO2 exhibits a 195.4-fold improvement in the CH4 yield compared to TiO2, accompanied by an increase in selectivity to 81%.
Copper Nanoclusters: Emerging Photoredox Catalysts for Organic Bond Formations
Arunachalam Sagadevan, Kathiravan Murugesan, Osman M. Bakr, Magnus Rueping
Chem. Commun., 2024, 60, 13858-13866
DOI: 10.1039/d4cc04774e
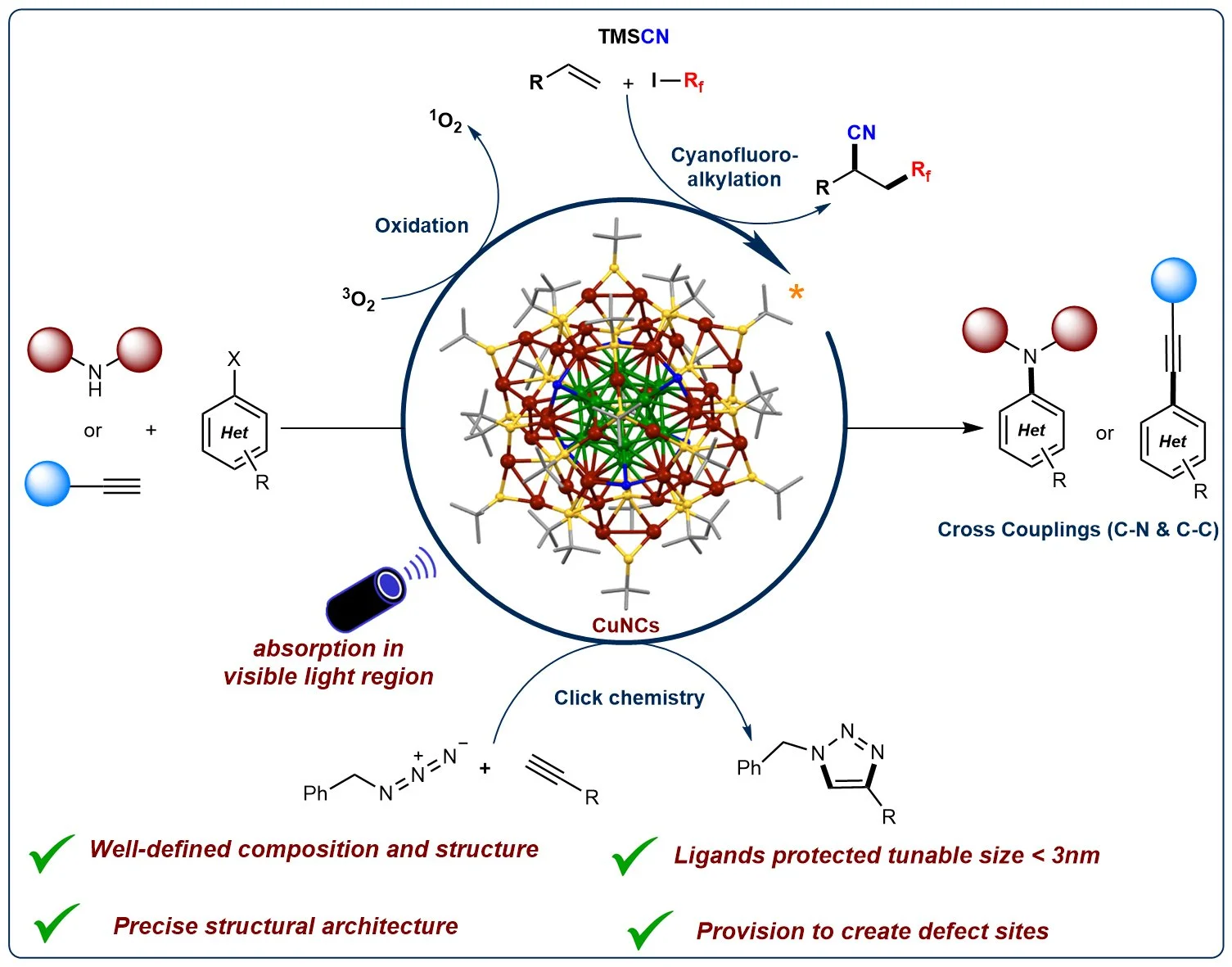
Abstract:
Advancements in fine chemical synthesis and drug discovery continuously demand the development of new and more efficient catalytic systems. In this regard, numerous transition metal-based catalysts have been developed and successfully applied in industrial processes. However, the need for innovative catalyst systems to further enhance the efficiency of chemical transformations and industrial applications persists. Metal nanoclusters (NCs) represent a distinct class of ultra-small nanoparticles (˂3 nm) characterized by a precise number of metal atoms coordinated with a defined number of ligands. This structure confers abundant unsaturated active sites and unique electronic and optical properties, setting them apart from conventional nanoparticles or bulk metals. The well-defined structure and monodisperse nature of NCs make them particularly attractive for catalytic applications. Among these, copper-based nanoclusters have emerged as versatile and sustainable catalysts for challenging organic bond-forming reactions. Their unique properties, including natural abundance, accessible oxidation states, diverse ligand architectures, and strong photophysical characteristics, contribute to their growing prominence in this field. In this article, we discuss the photocatalytic activities of Cu-based nanoclusters, focusing on their applications in cross-coupling reactions (C-C and C-N), click reactions, multicomponent couplings, and oxidation reactions.
Nian Li, Ruzal Sitdikov, Ajit Prabhakar Kale, Joost Steverlynck, Bo Li, Magnus Rueping
Beilstein J. Org. Chem., 2024, 20, 2500–2566.
DOI: 10.3762/bjoc.20.214

Abstract:
With the resurgence of electrosynthesis in organic chemistry, there is a significant increase in the number of routes available for late-stage functionalization (LSF) of drugs. Electrosynthetic methods, which obviate the need for hazardous chemical oxidants or reductants, offer unprecedented control of reactions through the continuous variation of the applied potential and the possibility of combination with photochemical processes. This capability is a substantial advantage for performing electrochemical or photoelectrochemical LSF. Ultimately, these protocols are poised to become a vital component of the medicinal chemist's toolkit. In this review, we discuss electrochemical protocols that have been demonstrated to be applicable for the LSF of pharmaceutical drugs, their derivatives, and natural substrates. We present and analyze representative examples to illustrate the potential of electrochemistry or photoelectrochemistry for the LSF of valuable molecular scaffolds.
Metallaphotoredox Catalysis for sp3 C–H Functionalizations through Single Electron Transfer (SET)
Jingchang Zhang, Magnus Rueping
Nature Catalysis, 2024, 7, 963-976
DOI: 10.1038/s41929-024-01215-3

Abstract:
Metallaphotoredox catalysis, which combines the strengths of photocatalysis and transition metal catalysis, has emerged as one of the most efficient platforms for sp³ C–H functionalization due to its exceptional ability to activate and transform inert bonds. In this process, photocatalysis typically facilitates the activation of C–H bonds to generate sp³-hybridized carbon-centered radicals, while the transition metal catalyst drives the subsequent transformation of these radicals into functionalized products. We highlight recent advances in sp³ C–H functionalizations achieved through metallaphotoredox catalysis, with a focus on photocatalytic single-electron transfer (SET) mechanisms, as distinct from hydrogen atom transfer (HAT) processes. The discussion begins by addressing the functionalization of different types of sp³ C–H bonds and then examines the role of various transition metal catalytic systems. We provide a comprehensive analysis of the diverse strategies employed in metallaphotoredox catalysis, along with their synthetic applications and mechanistic insights. Furthermore, we compare and contrast these strategies, aiming to inspire new reaction designs and drive further innovations in this rapidly evolving field.
Optimizing the reaction pathway of methane photo-oxidation over single copper sites
C. Feng, S. Zuo, M. Hu, Y. Ren, L., J. Luo, C. Zou, S. Wang, Y. Zhu, M. Rueping, Y. Han, H. Zhang
Nat.Commun. 2024, 15, 9088
DOI: 10.1038/s41467-024-53483-z
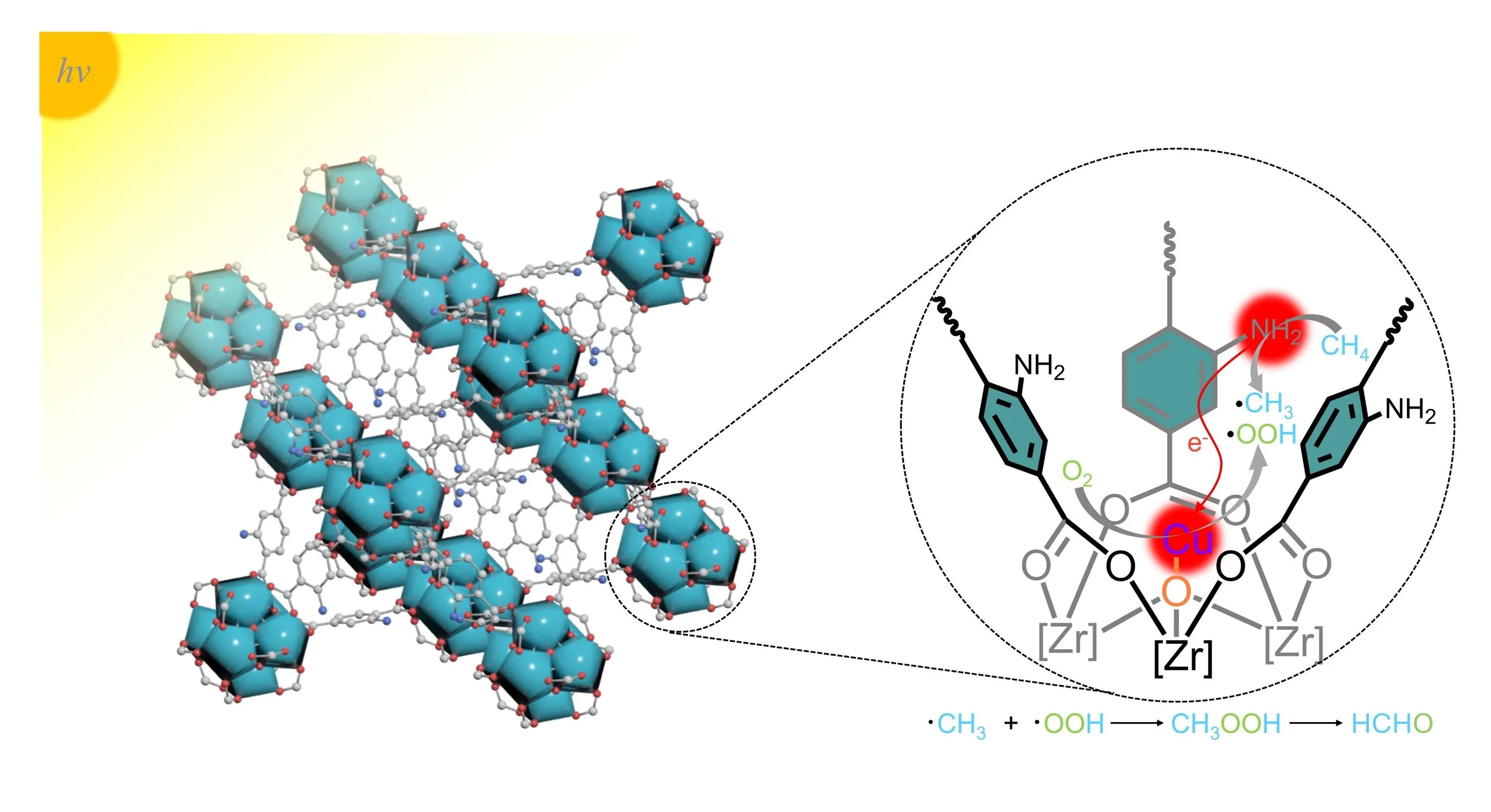
Abstract:
Direct photocatalytic conversion of methane to value-added C1 oxygenate with O2 is of great interest but presents a significant challenge in achieving highly selective product formation. Herein, a general strategy for the construction of copper single-atom catalysts with a well-defined coordination microenvironment is developed on the basis of metal-organic framework for selective photo-oxidation of CH4 to HCHO. We propose the directional activation of O2 on the mono-copper site breaks the original equilibrium and tilts the balance of radical formation almost completely toward •OOH. The synchronously generated •OOH and •CH3 radicals rapidly combine to form HCHO while inhibiting competing reactions, thus resulting in ultra-highly selective HCHO production (nearly 100%) with a time yield of 2.75 mmol gcat−1 h−1. This work highlights the potential of rationally designing reaction sites to manipulate reaction pathways and achieve selective CH4 photo-oxidation, and could guide the further design of high-performance single-atom catalysts to meet future demand.
Liang Yi, Deshen Kong, Ajit Prabhakar Kale, Bholanath Maity, Huifeng Yue, Amir Gizatullin, Rawan Alshehri, Rajesh Kancherla, Luigi Cavallo, Magnus Rueping
Angew. Chem. Int. Ed., 2024, 63, e202411961
DOI: 10.1002/anie.202411961

Abstract:
Bicyclo[1.1.1]pentane (BCP), recognized as a bioisostere for para-disubstituted benzene, has gained widespread interest in drug development due to its ability to enhance the physicochemical properties of pharmaceuticals. In this work, we introduce a photoinduced, halogen bonding-initiated, metal-free strategy for synthesizing various BCP derivatives. This method involves the generation of nucleophilic α-aminoalkyl radicals via halogen-bonding adducts. These undergo selective radical addition to [1.1.1]propellane, yielding electrophilic BCP radicals that subsequently participate in polarity-matched additions, culminating in the difunctionalization of bicyclopentane. The versatility and practicality of this metal-free approach are underscored by its broad substrate scope, which includes late-stage functionalization and a series of valuable transformations, all conducted under mild reaction conditions.
Mohammad Bodiuzzamana, Kathiravan Murugesana, Peng Yuana, Bholanath Maitya, Arunchalam Sagadevana, Naveen Halappaa, Song Wang, Partha Maity, Mohammed F. Alotaibi, De-en Jiang, Mutalifu Abulikemua, O. F. Mohammed, L. Cavallo, Osman M. Bakr, Magnus Rueping
J. Am. Chem. Soc. 2024, 146, 26994–27005
DOI: 10.1021/jacs.4c08688

Abstract:
Copper nanoclusters (Cu NCs) characterized by their well-defined electronic and optical properties are an ideal platform for organic photocatalysis and exploring atomic-level behaviors. However, their potential as greener, efficient catalysts for challenging reactions like decarboxylative oxygenation under mild conditions remains unexplored. Herein, we present Cu13(Nap)3(PPh3)7H10 (hereafter Cu13Nap), protected by 1-naphthalene thiolate (Nap), which performs well in decarboxylative oxidation (90% yield) under photochemical conditions. In comparison, the isostructural Cu13(DCBT)3(PPh3)7H10 (hereafter Cu13DCBT), stabilized by 2,4-dichlorobenzenethiolate (DCBT) and other previously reported Cu NCs (Cu28, Cu29, Cu45, Cu57, and Cu61) give the product in low yield. The introduction of naphthalene thiolate to the surface of Cu13 NCs influences the electronic structure and charge transfer in the ligand shell, enhancing visible light absorption and catalytic performance. Density functional theory (DFT) and experimental evidence suggest that the reaction proceeds primarily through an energy transfer mechanism. The energy transfer pathway is uncommon in the context of previous reports for decarboxylative oxidation reactions. Our findings suggest that strategically manipulating ligands holds significant potential for creating composite active sites on atomically precise copper NCs, resulting in enhanced catalytic efficacy and selectivity across various challenging reactions.
Nickel-Catalyzed Selective Disulfides Formation by Reductive Cross-Coupling of Thiosulfonates
Tingting Yuan, Xiang-Yu Chen, Tengfei Ji, Huifeng Yue, Kathiravan Murugesan, Magnus Rueping
Chem. Sci. 2024, 15, 15474–15479
DOI: 10.1039/D4SC02969K

Abstract:
Developing a new methodology to prepare disulfides is of importance in modern organic chemistry. Despite the rapid progress in nickel-catalyzed reductive cross-coupling reactions for forming carbon-carbon/heteroatom bonds, synthesizing S-S bonds has posed a significant challenge. We report using nickel catalysts for the reductive cross-coupling of thiosulfonates. This method operates under mild conditions and provides a convenient pathway to synthesize a range of unsymmetrical and symmetrical disulfides from readily available, bench-stable thiosulfonates with excellent selectivity. Importantly, this approach can be applied to modify pharmaceuticals at later stages and prepare various targeted compounds.
Y. Li, C. Chen, Z. Cao, G. Zhao, S. Zuo, Z. Wu, Z. Wang, L. Zheng, Z. Lai, M. Rueping, Y. Han, H. Zhang
Angew. Chem. Int. Ed., 2024, 63, e202411218
DOI: 10.1002/anie.202411218

Abstract:
Chemical modification via functional dopants in carbon materials holds great promise for elevating catalytic activity and stability. To gain comprehensive insights into the pivotal mechanisms and establish structure-performance relationships, especially concerning the roles of dopants, remains a pressing need. Herein, we employ computational simulations to unravel the catalytic function of heteroatoms in the acidic oxygen evolution reaction (OER), focusing on a physical model of high-electronegative F and N co-doped carbon matrix. Theoretical and experimental findings elucidate that the enhanced activity originates from the F and pyridinic-N (Py-N) species that achieve carbon activation. This activated carbon significantly lowers the conversion energy barrier from O* to OOH*, shifts the potential-limiting step from OOH* formation to O* generation, and ultimately optimizes the energy barrier of the potential-limiting step. This wok elucidates that the critical role of heteroatoms in catalyzing the reaction and unlocks the potential of carbon materials for acidic OER.
Tracking Water Splitting Activity by Cocatalyst Identity in SrTiO3
Nursaya Zhumabay, Jeremy A. Bau, Rafia Ahmad, Laurentiu Braic, Huabin Zhang, Luigi Cavallo, Magnus Rueping
Small Struct. 2024, 2400283
DOI: 10.1002/sstr.202400283

Abstract:
Photocatalytic water splitting is the most idealistic route to green hydrogen production, but the extensive material requirements for this reaction make it difficult to realize good photocatalysts. Noble metal cocatalysts are often added to photocatalysts to aid in charge separation and improve surface kinetics for H2 evolution. In this study, the high activity of the promising photocatalyst Al-doped SrTiO3 is demonstrated to be ultimately dependent on the cocatalyst used as much as the presence of Al dopant. By tracking the band energetics of photocatalyst electrodes using operando electrochemical ATR-SEIRAS, cocatalysts (especially Rh) are found to shift the quasi-Fermi levels and metal-semiconductor flat-band potentials of photocatalysts in an anodic direction. Furthermore, the size of the shift directly correlates with overall water splitting activity, demonstrating that SrTiO3 becomes more active as photo-generated electrons are stabilized further from the conduction band. Rh on Al-doped SrTiO3 provides the most advantageous band tailoring as confirmed by DFT, and is experimentally found to provide this effect by eliminating Ti3+-related surface traps in the presence of Al dopants. Therefore, the effect of cocatalysts on water splitting activity is more complicated than previously thought.
V. K. Velisoju, E. V. Ramos-Fernandez, R. Kancherla, K. Pal, H. Mohamed, J. L. Cerrillo, R. Ahmad, M. Meijerink, L. Cavallo, M. Rueping, P. Castaño
Angew. Chem. Int. Ed., 2024, 63, e202409490
DOI: 10.1002/anie.202409490

Abstract:
Our study unveils a pioneering methodology that effectively distributes Pd species within a zeolitic imidazolate framework-8 (ZIF-8). We demonstrate that Pd can be encapsulated within ZIF-8 as atomically dispersed Pd species that function as an excited-state transition metal catalyst for promoting carbon–carbon (C–C) cross-couplings at room temperature using visible light as the driving force. Furthermore, the same material can be reduced at 250 °C, forming Pd metal nanoparticles encapsulated in ZIF-8. This catalyst shows high rates and selectivity for carbon dioxide hydrogenation to methanol under industrially relevant conditions (250 °C, 50 bar): 7.46 molmethanol molmetal−1 h−1 and >99%. Our results demonstrate the correlations of the catalyst structure with the performances at experimental and theoretical levels.
Kamila Almagambetova, Kathiravan Murugesan, Magnus Rueping
ACS Catal., 2024, 14, 12664−12670
DOI: 10.1021/acscatal.4c03766
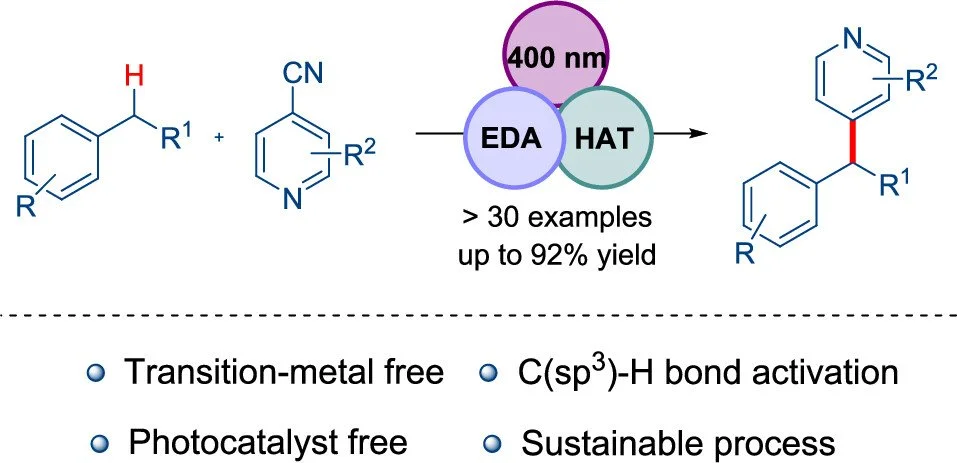
Abstract:
Developing a transition-metal-free system for functionalizing the C(sp3)–H bond has posed a long-standing challenge in synthetic organic chemistry and is of significance in drug discovery. In this context, we present a straightforward protocol for the heteroarylation of benzylic and alpha-amino C–H bonds, achieved without transition metals or photocatalysts. A pivotal aspect of redox-neutral hydroarylation is the utilization of commercially available triisopropylsilanethiol (iPr3SiSH) as an organocatalyst. This organocatalyst serves a dual function in forming an electron donor–acceptor complex (EDA) and acting as a hydrogen atom transfer catalyst, generating alkyl/benzylic radicals from the corresponding C–H bonds. Applying this mild methodology, various benzylic and amino C–H bonds were functionalized to give a C(sp3)–H and C(sp2)–heteroaryl C–C cross-coupled product. To demonstrate the practical utility of this protocol, pyridine was incorporated into a drug molecule via late-stage modification. Additionally, a gram-scale reaction was also performed to show the advantages of this protocol.
Huawei Huang, Liangliang Xu, Shouwei Zuo, Lu Song, Chen Zou, Max García-Melchor, Yang Li, Yuafu Ren, Magnus Rueping, Huabin Zhang
Adv. Mater. 2024, 2405128
DOI: 10.1002/adma.202405128

Abstract:
In alkaline water electrolysis and anion exchange membrane water electrolysis technologies, the hydrogen evolution reaction (HER) at the cathode is significantly constrained by a high energy barrier during the water dissociation step. This study employs a phase engineering strategy to construct heterostructures composed of crystalline Ni4W and amorphous WOx aiming to enhance catalytic performance in the HER under alkaline conditions. This work systematically modulates the oxidation states of W within the amorphous WOx of the heterostructure to adjust the electronic states of the phase boundary, the energy barriers associated with the water dissociation step, and the adsorption/desorption properties of intermediates during the alkaline HER process. The optimized catalyst, Ni4W/WOx-2, with a quasi-metallic state of W coordinated by a low oxygen content in amorphous WOx, demonstrates exceptional catalytic performance (22 mV@10 mA cm−2), outperforming commercial Pt/C (30 mV@10 mA cm−2). Furthermore, the operando X-ray absorption spectroscopy analysis and theoretical calculations reveal that the optimized W atoms in amorphous WOx serve as active sites for water dissociation and the nearby Ni atoms in crystalline Ni4W facilitated the release of H2. These findings provide valuable insights into designing efficient heterostructured materials for energy conversion.
Visible-Light-Induced Excited-State Copper Catalysis: Recent Advances and Perspectives
Nian Li, Bo Li, Kathiravan Murugesan, Arunachalam Sagadevan, Magnus Rueping
ACS Catal., 2024, 14, 11974-11989
DOI: 10.1021/acscatal.4c03238
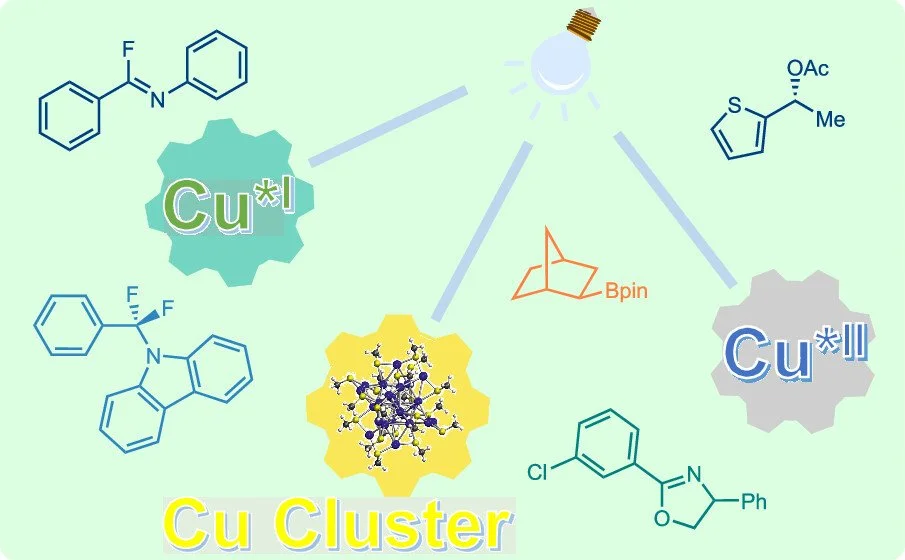
Abstract:
Photoactive copper complexes have gained significant attention due to their photocatalytic activities. Different homogeneous Cu(I) complexes, Cu(II) complexes, and heterogeneous copper-based photocatalysts have been investigated and utilized in a broad spectrum of organic transformations. These applications span radical additions, C–C bond and C–heteroatom bond cross-couplings, aerobic oxidative reactions, and kinetic resolutions. This review summarizes the advancements in this dynamic field of visible-light-induced, excited-state copper-catalyzed organic reactions over recent years. It is organized according to the type of excited copper species involved and provides a perspective on current and future developments.
Alice Nanni, Deshen Kong, Chen Zhu, Magnus Rueping
Green Chem., 2024, 26, 8341-8347
DOI: 10.1039/D4GC01790K
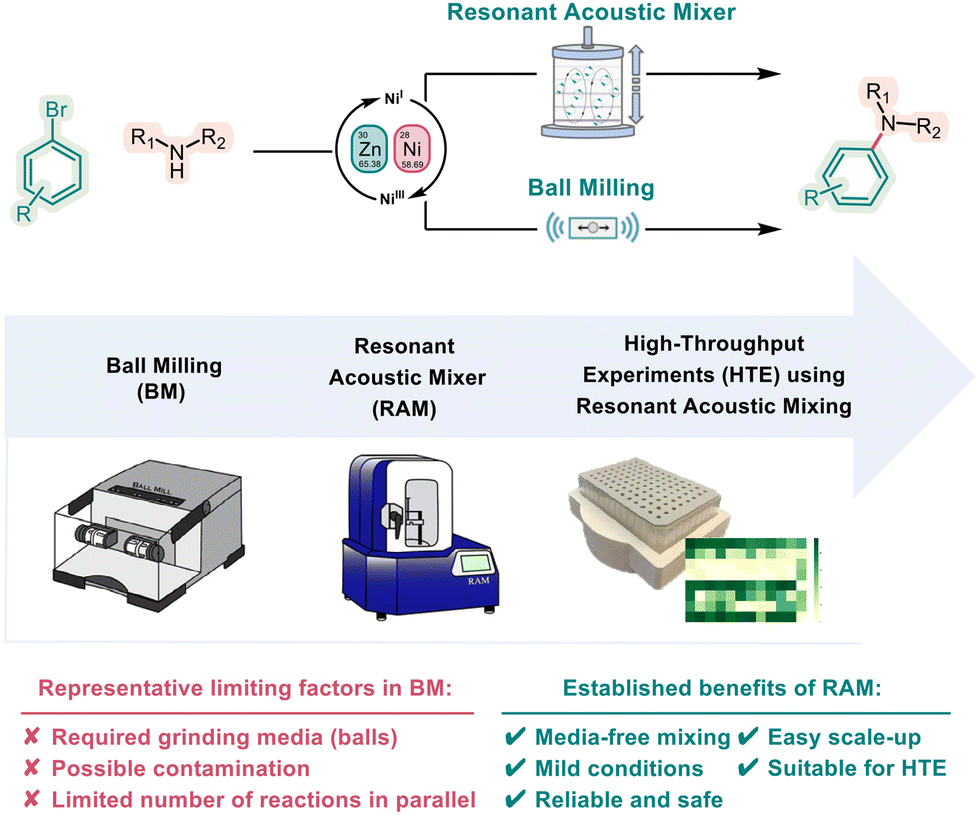
Abstract:
In recent years, mechanochemistry has become recognized as an efficient, practical, and sustainable alternative for chemical synthesis. Adhering to the principles of green chemistry, mechanochemistry enables solvent-free, faster, and energy-efficient reactions, thereby reducing waste production and enhancing atom economy. Herein, we present a new nickel-catalyzed mechanochemical High Throughput Experimentation (HTE) amination protocol enabled by Resonant Acoustic Mixing (RAM). The developed C–N cross-coupling reactions avoid possible contamination, scaling-up challenges, and parallel reaction limitations by applying an accelerated screening and optimization protocol. The reduced amount of solvents in the reactions and the minimal amount of reagents required highlight the advantages of our approach over most common solvent-based reactions, aligning with the principles of sustainability and resource efficiency. Furthermore, the mechanochemistry methodology demonstrates seamless scalability to a multigram scale without additional optimizations, emphasizing its potential for streamlined, environmentally friendly, and large-scale industrial production.
Ram Karan, Dominik Renn, Thorsten Allers and Magnus Rueping
Front. Microbiol., 15, 1403623
DOI: 10.3389/fmicb.2024.1403623

Abstract:
Extremophilic proteins are valuable in various fields, but their expression can be challenging in traditional hosts like Escherichia coli due to misfolding and aggregation. Haloferax volcanii (H. volcanii), a halophilic expression system, offers a solution. This study examined cleavable and non-cleavable purification tags at both the N- and C-termini when fused with the superfolder green fluorescent protein (sfGFP) in H. volcanii. Our findings reveal that an N-terminal 8xHis-tag or Strep-tag®II significantly enhances protein production, purity, and yield in H. volcanii. Further experiments with mCherry and halophilic alcohol dehydrogenase (ADH) showed improved expression and purification yields when the 8xHis-tag or Strep-tag®II was positioned at the C-terminus for mCherry and at the N-terminus for ADH. Co-positioning 8xHis-tag and Twin-Strep-tag® at the N-terminus of sfGFP, mCherry, and ADH yielded significantly enhanced results. These findings highlight the importance of thoughtful purification tag design and selection in H. volcanii, providing valuable insights for improving protein production and purification with the potential to advance biotechnological applications.
Badriah Alamer, Arunachalam Sagadevan, Mohammad Bodiuzzaman, Kathiravan Murugesan, Salman Alsharif, Ren-Wu Huang, Atanu Ghosh, Malenahalli H. Naveen, Chunwei Dong, Saidkhodzha Nematulloev, Jun Yin, Aleksander Shkurenko, Mutalifu Abulikemu, Xinglong Dong, Yu Han, Mohamed Eddaoudi, Magnus Rueping, and Osman M. Bakr
J. Am. Chem. Soc., 2024, 146, 23, 16295–16305
DOI: 10.1021/jacs.4c05077
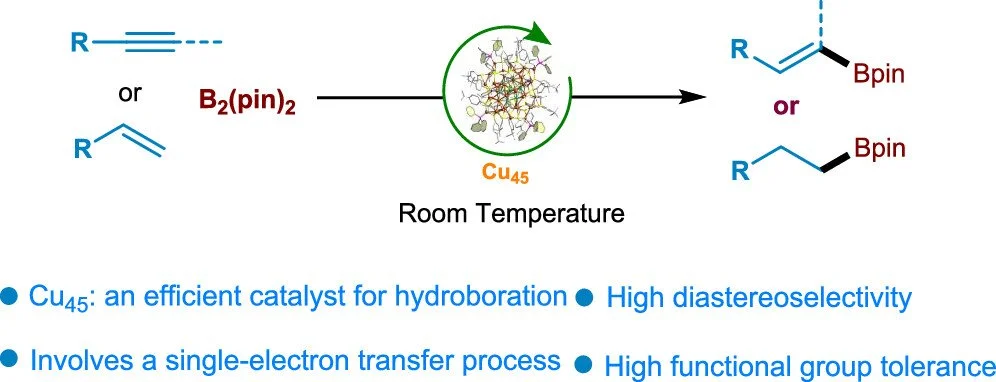
Abstract:
Atomically precise metal nanoclusters (NCs) have become an important class of catalysts due to their catalytic activity, high surface area, and tailored active sites. However, the design and development of bond-forming reaction catalysts based on copper NCs are still in their early stages. Herein, we report the synthesis of an atomically precise copper nanocluster with a planar core and unique shell, [Cu45(TBBT)29(TPP)4(C4H11N)2H14]2+ (Cu45) (TBBT: 4-tert-butylbenzenethiol; TPP: triphenylphosphine), in high yield via a one-pot reduction method. The resulting structurally well-defined Cu45 is a highly efficient catalyst for the hydroboration reaction of alkynes and alkenes. Mechanistic studies show that a single-electron oxidation of the in situ-formed ate complex enables the hydroboration via the formation of boryl-centered radicals under mild conditions. This work demonstrates the promise of tailored copper nanoclusters as catalysts for C–B heteroatom bond-forming reactions. The catalysts are compatible with a wide range of alkynes and alkenes and functional groups for producing hydroborated products.
Eswaravara Prasadarao Komarala, Aasif Asharafbhai Dabbawala, Messaoud Harfouche, Michalis A. Vasiliades, Nikolaos Charisiou, Dalaver H. Anjum, Samuel Mao, Magnus Rueping, Mark A. Baker, Maria A. Goula, Angelos M. Efstathiou, Kyriaki Polychronopoulou
Appl. Surf. Sci., 2024, 160388
DOI: 10.1016/j.apsusc.2024.160388.

Abstract:
Dry reforming of methane (DRM) is an inimitable approach for eliminating both greenhouse gases, methane and CO2, while producing synthesis gas which further can be converted to added-value fuels. However, the industrialization of DRM has been limited due to sintering and coking to the catalysts under the harsh reaction conditions. Here, we propose a new methodology for producing tunable supported nickel-based catalysts for DRM using metal organic frameworks (MOFs) as precursors of the catalyst components. In particular, Ni- and La-MOFs were coalesced using sonication and then calcined at different temperatures to produce catalysts with tailored Ni metal size (5–20 nm) and tuned strong metal-support interactions (SMSI). Here, the synthesis of nickel and nickel oxide nanoparticles embedded on lanthanum-based supports by controlled calcination of the MOF structures is demonstrated; the Ni supported catalysts were then used for DRM reaction. Among the developed catalysts, the catalyst produced following calcination at 500 °C (Ni-La-500) has shown stable and high CH4 conversion rates under DRM conditions at 800 °C with negligible carbon formation at elevated gas hourly space velocities. Further, the catalytic performance of Ni-La-500 catalyst (sonicated) was compared to the physically mixed MOFs derived catalyst (Ni-La-500-PM), conventional wetness impregnated catalyst (Ni-La-500 WI), Ni-MOF derived unsupported catalyst (Ni-500), and lower Ni concentration catalyst (sonicated, 10Ni-La-500). The observed CH4 conversion rates at 50,000 mL·gcat–1·h−1 GHSV are 81.8, 67.9, 85.4, 34.7, and 57.9 % for Ni-La-500, Ni-La-500 PM, Ni-La-500 WI, Ni-500, and 10Ni-La-500 catalysts, respectively after 24 h of DRM reaction. Overall, with the herein proposed approach of MOF-derived supported catalysts exceptional conversion rates and stability during the DRM reaction with nominal coking and sintering were demonstrated, solving the two major challenges faced by conventional and unsupported catalysts.
Nickel-catalyzed C(sp2)–C(sp3) coupling via photoactive electron donor–acceptor (EDA) complexes
Salman Alsharif, Chen Zhu, Xiushan Liu, Shao-Chi Lee, Huifeng Yue, Magnus Rueping
Chem. Commun., 2024, 5153–5156
DOI: 10.1039/D4CC00217B

Abstract:
We have developed a novel Ni-catalyzed reductive cross-coupling reaction of aryl bromides and alkyl iodides via a photoactive electron donor–acceptor (EDA) complex. This photo-induced process enables the efficient construction of C(sp2)–C(sp3) bonds in the absence of an external photocatalyst. Electronically and structurally diverse aryl bromides, as well as secondary and primary alkyl iodides could undergo this transformation smoothly. Natural product derivatives were employed successfully, and UV-vis spectroscopy was utilized to gain mechanistic insight.
Multiphoton photoredox catalysis enables selective hydrodefluorinations
Jiaqi Jia, Kathiravan Murugesan, Chen Zhu, Huifeng Yue, Shao-Chi Lee, Magnus Rueping
Chinese Chemical Letters, 2024, 109866
DOI: 10.1016/j.cclet.2024.109866
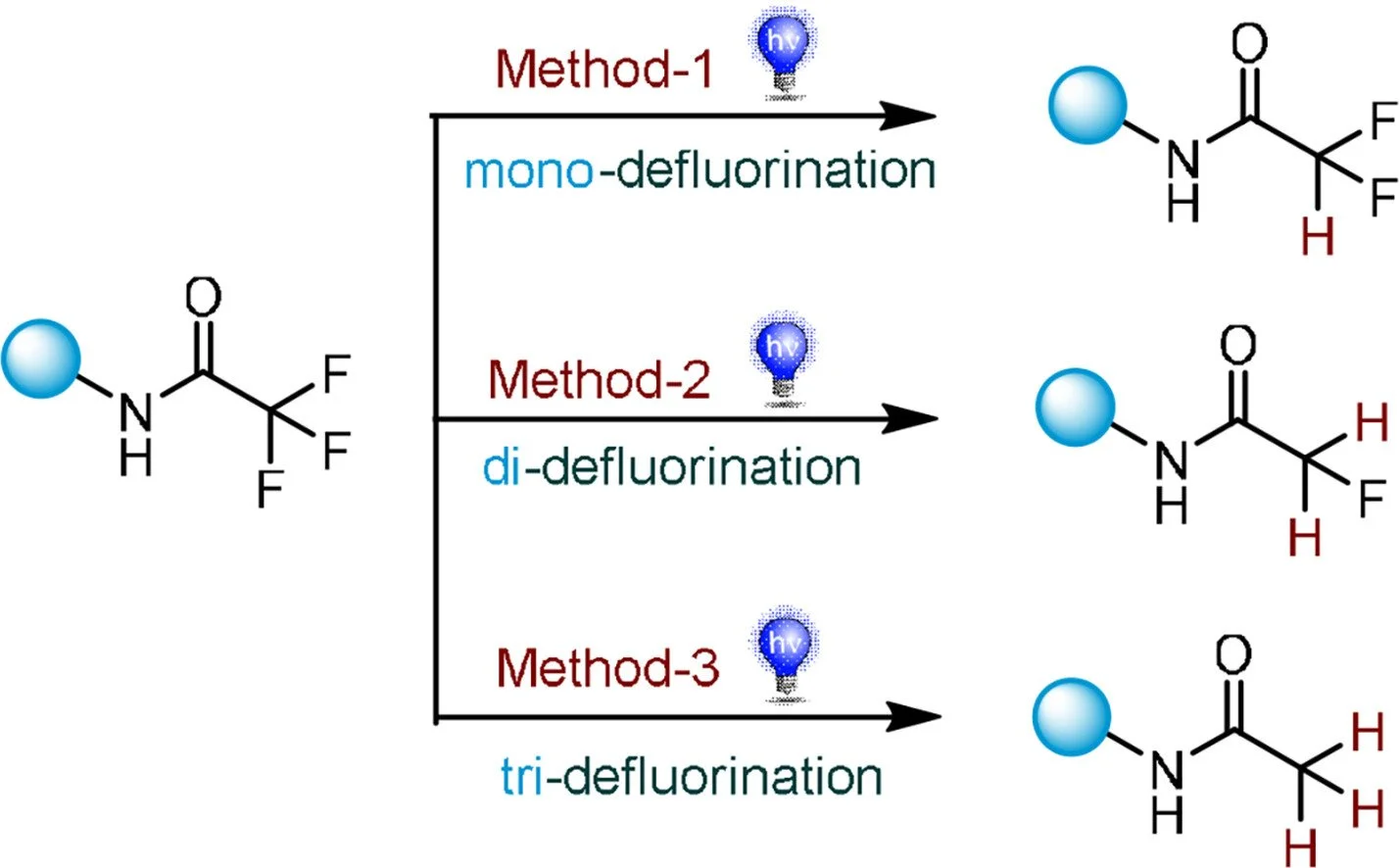
Abstract:
Late-stage modification of complex molecules via site-selective hydrodefluorination is a challenging endeavor. The selective activation of carbon-fluorine (C–F) bonds in the presence of multiple C–F bonds is of importance in organic synthesis and drug discovery. Herein, we describe the activation of C-F bonds via multiphoton photoredox catalysis to selectively produces a series of hydrodefluorinated compounds by simply tuning the reaction conditions. Moreover, this protocol was successfully applied to the late-stage functionalization of different drug-derivatives and the corresponding mono-, di-, and tri-defluorinated products were obtained in good to excellent yields. A detailed mechanistic investigation provides insight into the unprecedented hydrodefluorination pathway.
Jiaqi Jia, Serik Zhumagazy, Chen Zhu, Shao-Chi Lee, Salman Alsharif, Huifeng Yue, Magnus Rueping
Chem. Eur. J., 2024, e202302927
DOI: 10.1002/chem.202302927

Abstract:
A new cross-coupling of trifluoromethyl arenes has been realized via multiphoton photoredox catalysis. Trifluoromethyl arenes were demonstrated to undergo selective mono-defluorinative alkylation under mild reaction conditions providing access to a series of valuable α,α-difluorobenzylic compounds. The reaction shows broad substrate scope and general functional group tolerance. In addition to the electron-deficient trifluoromethyl arenes that are easily reduced to the corresponding radical anion, more challenging electron-rich substrates were also successfully applied. Steady-State Stern-Volmer quenching studies indicated that the trifluoromethyl arenes were reduced by the multiphoton excited Ir-based photocatalyst.
Atanu Ghosh, Arunachalam Sagadevan, Kathiravan Murugesan, Stefan Adrian F. Nastase, Bholanath Maity, Mohammad Bodiuzzaman, Aleksander Shkurenko, Mohamed Nejib Hedhili, Jun Yin, Omar F. Mohammed, Mohamed Eddaoudi, Luigi Cavallo, Magnus Rueping, Osman M. Bakr
Mater. Horiz., 2024, 11, 2494-2505
DOI: 10.1039/D4MH00098F
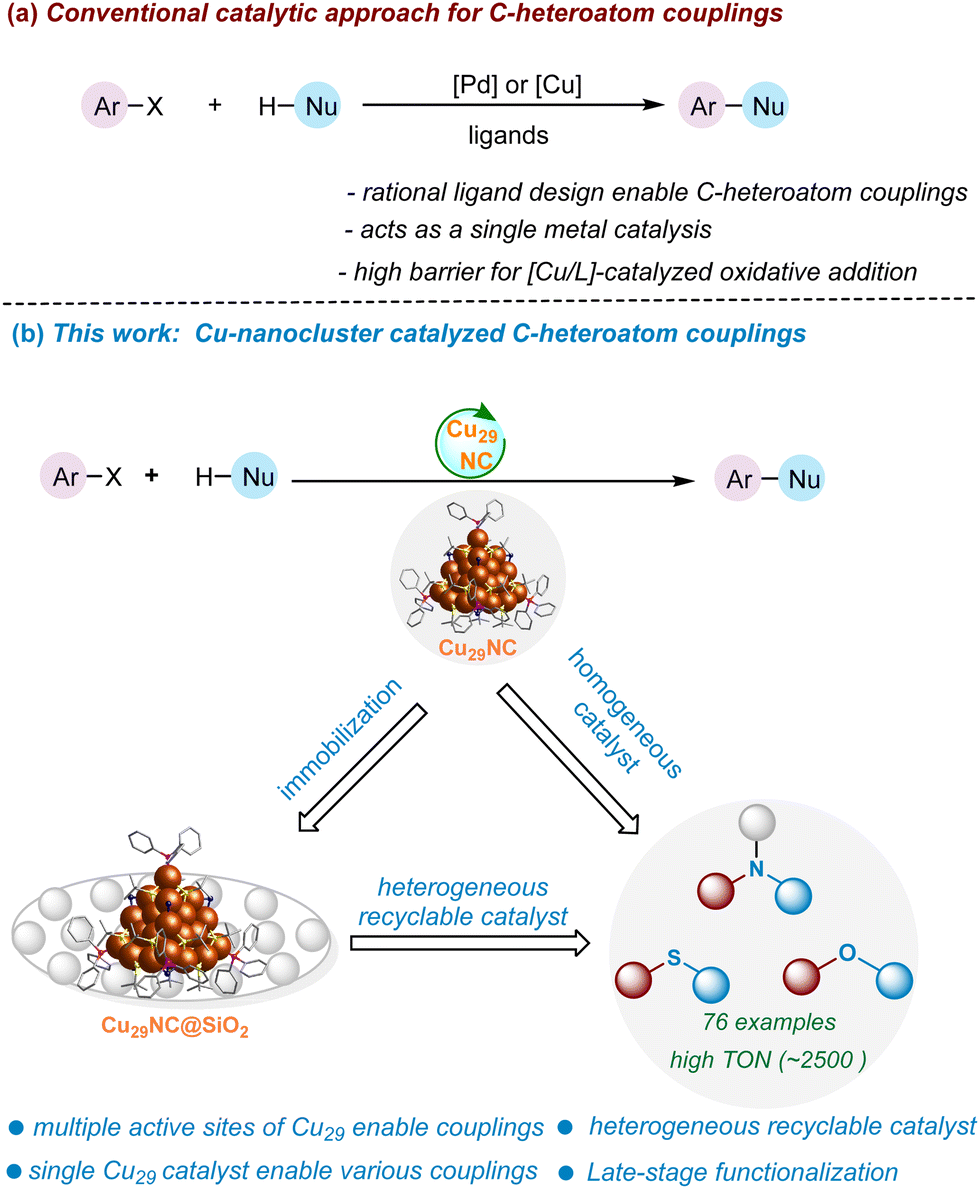
Abstract:
Atomically precise copper nanoclusters (NCs) are an emerging class of nanomaterials for catalysis. Their versatile core–shell architecture opens the possibility of tailoring their catalytically active sites. Here, we introduce a core–shell copper nanocluster (CuNC), [Cu29(StBu)13Cl5(PPh3)4H10]tBuSO3 (StBu: tert-butylthiol; PPh3: triphenylphosphine), Cu29NC, with multiple accessible active sites on its shell. We show that this nanocluster is a versatile catalyst for C-heteroatom bond formation (C–O, C–N, and C–S) with several advantages over previous Cu systems. When supported, the cluster can also be reused as a heterogeneous catalyst without losing its efficiency, making it a hybrid homogeneous and heterogeneous catalyst. We elucidated the atomic-level mechanism of the catalysis using density functional theory (DFT) calculations based on the single crystal structure. We found that the cooperative action of multiple neighboring active sites is essential for the catalyst's efficiency. The calculations also revealed that oxidative addition is the rate-limiting step that is facilitated by the neighboring active sites of the Cu29NC, which highlights a unique advantage of nanoclusters over traditional copper catalysts. Our results demonstrate the potential of nanoclusters for enabling the rational atomically precise design and investigation of multi-site catalysts.
Prashant S. Shinde, Valmik S. Shinde, Magnus Rueping
Chem. Commun., 2024, 60, 3826
DOI: 10.1039/d4cc00309h
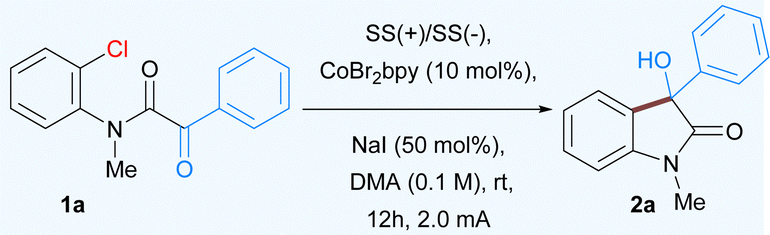
Abstract:
The development of an electrochemical cobalt catalyzed C–Cl bond activation at room temperature for the nucleophilic addition of aryl and vinyl chlorides to α-ketoamides is described. The overall method operates through an electrochemically induced low valent cobalt catalyst that oxidatively adds to aryl or vinyl chlorides affording medicinally important 3-hydroxy oxindole and 3-hydroxypyrrolidinone scaffolds. The development of an enantioselective version using a chiral pyrox ligand is also demonstrated.
Lidia Karpova, Matěj Daniel, Rajesh Kancherla, Krishnamoorthy Muralirajan, Bholanath Maity, Magnus Rueping
Org. Lett., 2024, 26, 1657–1661
DOI: 10.1021/acs.orglett.4c00147

Abstract:
Excited-state nickel-catalyzed C–N cross-coupling of aryl bromides with sodium azide enables the synthesis of diarylamines and primary anilines under mild reaction conditions. The oxidative addition of electron-rich aryl bromides with low-valent Ni under the photochemical conditions is endothermic. Herein, we demonstrate a light-mediated nickel-catalyzed reaction of electronically rich aryl bromides that yields diarylamines, while the reaction with electron-deficient aryl bromides gives access to anilines at room temperature. Overcoming the limitations associated with the activation of electronically rich aryl bromides in photochemical nickel catalysis, we developed a novel photoinduced Ni-catalyzed C–N cross-coupling reaction involving aryl bromides and NaN3 at room temperature. A wide range of aryl bromides, including both EDGs and EWGs, were effectively cross-coupled with sodium azide to give diarylamines and anilines in good yields. A mechanism involving the NiI/NiIII catalytic pathway in which the photoreduction of NiII to NiI is crucial for C–N cross-coupling is proposed.
Boosted hydrogen evolution kinetics of heteroatom-doped carbons with isolated Zn as an accelerant
Yang Li, Shouwei Zuo, Fen Wei, Cailing Chen, Guikai Zhang, Xiaojuan Zhao, Zhipeng Wu, Jing Zhang, Sibo Wang, Wei Zhou, Magnus Rueping, Yu Han, Huabin Zhang
PNAS, 2024, 121, e2315362121
DOI: 10.1073/pnas.2315362121

Abstract:
Carbon- based single- atom catalysts, a promising candidate in electrocatalysis, offer insights into electron- donating effects of metal center on adjacent atoms. Herein, we present a practical strategy to rationally design a model catalyst with a single zinc (Zn) atom coordinated with nitrogen and sulfur atoms in a multilevel carbon matrix. The Zn site exhibits an atomic interface configuration of ZnN4S1, where Zn's electron injection effect enables thermal- neutral hydrogen adsorption on neighboring atoms, pushing the activity boundaries of carbon electrocatalysts toward electrochemical hydrogen evolution to an unprecedented level. Experimental and theoretical analyses confirm the low- barrier Volmer–Tafel mechanism of proton reduction, while the multishell hollow structures facilitate the hydrogen evolution even at high current intensities. This work provides insights for understanding the actual active species during hydrogen evolution reaction and paves the way for designing high- performance electrocatalysts.
Chao-Shen Zhang, Chang-Zhen Fang, Liang Yi, Chen Zhu, c Zhi-Xiang Wang, Xiang-Yu Chen and Magnus Rueping
Org. Chem. Front., 2024, 11, 673-683
DOI: 10.1039/D3QO01779F

Abstract:
Visible light photoredox-catalyzed reactions have become essential tools in organic synthesis. However, these transformations often rely on high-cost photocatalysts that require multistep synthesis and chromophore design. In recent years, charge transfer complex (CTC) photochemistry has emerged as an alternative to photoredox catalysis, but this method requires substrates to form the CTC. To overcome these limitations, we developed a new catalytic paradigm using easily available N-heterocyclic nitreniums (NHNs) as photocatalysts. Our method uses incident light absorbed by the in situ-formed CTC between NHN and its counteranion or a sacrificial donor, extending CTC photochemistry to a general catalytic mode without relying on substrates. Here, we demonstrate NHN catalysis for the photoreduction of C–Cl bonds in chloroform and activated alkyl chlorides. This approach is characterized by easily available NHNs (one-step synthesis without column purification), operational simplicity, and diverse transformations. Mechanistic studies reveal that the generated NHN aminyl radical could undergo inner-sphere single-electron transfer in concert with halogen-atom dissociation.
Rajesh Kancherla, Krishnamoorthy Muralirajan, Sayan Dutta, Kuntal Pal, Bo Li, Bholanath Maity, Luigi Cavallo, Magnus Rueping
Angew. Chem. Int. Ed., 2024, 63, e202314508
DOI: 10.1002/anie.202314508

ABSTRACT:
The development of metal complexes that function as both photocatalyst and cross-coupling catalyst remains a challenging research topic. So far, progress has been shown in palladium(0) excited-state transition metal catalysis for the construction of carbon-carbon bonds where the oxidative addition of alkyl/aryl halides to zero-valent palladium (Pd0) is achievable at room temperature. Distinct from Pd0-complexes that easily undergo oxidative addition with aryl/alkyl halides, the oxidative addition of aryl halides to electrophilic divalent Pd(II) is uphill and energy-demanding. Overcoming these limitations, we realized the oxidative addition of aryl halides to divalent palladium using mild photochemical conditions at room temperature under open air conditions. Various electron withdrawing and donating aryl/heteroaryl iodides were cross-coupled with sodium azide to give primary anilines by using Pd(OAc)2 as both photocatalyst and cross-coupling catalyst. We demonstrate for the first time, that divalent PdII can act as a light-absorbing species which undergoes double excitation to give the C−N cross-coupled product under air. DFT studies and mechanistic investigations support the photoexcitation of two distinct divalent palladium species and unravel the reaction mechanism.