
Kuntal Pal, Rajesh Kancherla, Sayan Dutta, Serik Zhumagazy, Bholanath Maity, Luigi Cavallo, Magnus Rueping
Angew. Chem. Int. Ed. 2025, e22979
DOI: 10.1002/anie.202522979
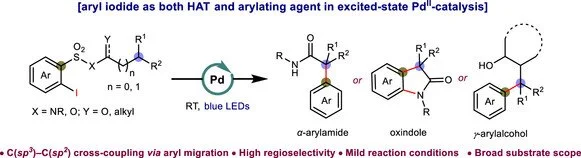
Abstract:
Catalytic modification of C−H bonds using palladium is highly appealing, but transformations involving alkyl-Pd(II) intermediates with β-hydrogens are challenging due to the propensity for β-hydride elimination. To address this, selective arylation of sp3 C−H bonds in amides and alcohols has been achieved using divalent palladium as an excited-state transition metal (TM) species at room temperature. Our approach unveils a non-redox-neutral pathway to achieve C(sp3)–C(sp2) cross-couplings, utilizing aryl iodides as both hydrogen atom transfer (HAT) and arylating agents for the first time under Pd-photochemical conditions. The mechanism involves photo-induced Pd(II)-catalyzed aryl radical generation, HAT, and aryl migration, effectively bypassing the common β-hydride elimination and annulation pathways. Mechanistic investigations using isotope-labeled compounds and density functional theory (DFT) studies support the mechanistic rationale, affirming both the feasibility and intricacies of this effective transformation.
Gas–Proton Microenvironment Modulation for Enhanced CO2-to-Formate Electroreduction
Jifeng Wu, Yu Li, Miao Hu, Tingsong Li, Moyu Yi, Xiangyun Xiao, Honglin Li, Yongfeng Guo, Yoji Kobayashi, Magnus Rueping, Wan-Lu Li, Huabin Zhang, Kuo-Wei Huang
Angew. Chem. Int. Ed. 2025, e202516163
DOI: 10.1002/anie.202516163
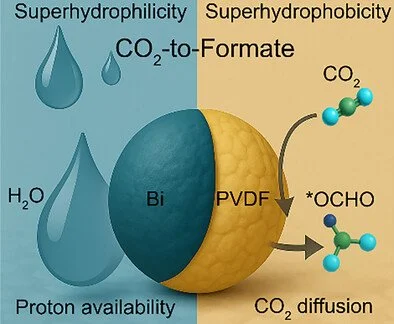
Abstract:
Precise control of interfacial water structure is essential for suppressing side reactions and enabling selective CO2 electroreduction at industrial current densities. Here, we synthesize a series of bismuth-based catalysts with spatially encoded superhydrophilic–superhydrophobic nanodomains by partially embedding polyvinylidene fluoride (PVDF) into Bi nanoparticles. This strategy creates interfacial polarity patterns that stabilize *OCHO intermediates while suppressing hydrogen and CO evolution. Compared to the PVDF-free control, the optimized Bi–PVDF catalyst exhibits significantly enhanced formate partial current density, Faradaic efficiency (FE), and long-term stability. It achieves > 90% FE at −200 mA cm−2 for 50 h and maintains high selectivity up to −700 mA cm−2. Operando spectroscopy and multiscale simulations reveal that the dual-wettability interface modulates local hydration and charge distribution, promoting selective intermediate formation while kinetically suppressing side pathways. By addressing the longstanding challenge of coupled gas–proton transport, this work offers a mechanism-driven and scalable strategy to construct interfacial microenvironments for high-rate, selective CO2 electroreduction.
Rajesh Kumar Parsapur, Georgian Melinte, Youssef Saih, Edy Abou-Hamad, Vasilios Samaras, Robert Peter Hodgkins, Omer Refa Koseoglu, Mohammad Fuad Aljishi, Anissa Bendjeriou-Sedjerari, Zhiping Lai, Magnus Rueping, Kuo-Wei Huang
J. Am. Chem. Soc. 2025, 147, 44, 41109–41121
DOI: 10.1021/jacs.5c15691
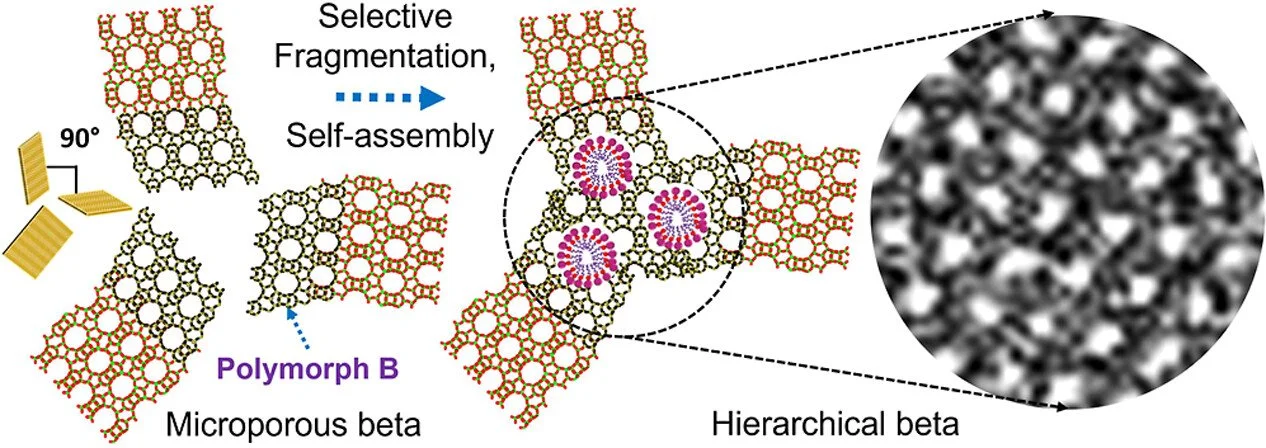
Abstract:
Designing hierarchical zeolites with ordered mesoporosity remains a major synthetic challenge despite their well-established importance in catalysis and separations. In this study, we examine the spatiotemporal dynamics of zeolite dissolution and reorganization to retrace how transient structural changes during zeolite nucleation influence supramolecular self-assembly. For the first time, we demonstrate that subtle differences in the aluminum (Al) spatial proximity and distribution within various zeolite frameworks critically affect their mesostructuring behavior. Notably, beta zeolite comprising distinct polymorphs with marginally different Al environments has shown an anisotropic dissolution pattern, where polymorph B was selectively fragmented, leaving polymorph A unaffected. Through a postsynthetic mesostructuring approach, polymorph B was preferentially reorganized into a gyroidal cubic ordered mesophase, resulting in partially ordered mesoporous beta zeolite. The prepared zeolites exhibit improved catalytic performance in vacuum gas oil hydrocracking, affording high isoparaffin yields and superior naphtha selectivity. These findings provide a direct link between atomic-scale heterogeneity and mesoscale structural evolution in crystals, offering a new strategy for designing advanced catalytic materials.
Selective Electrocatalytic CO2 Reduction to Methanol: A Roadmap toward Practical Implementation
Abdulrahman Allangawi, Xiangyun T. Xiao, Xiao Ma, Mayasem Alsuhami, Mohd Adnan Khan, Rashed Aleisa, Yoji Kobayashi, Wan-Lu Li, Magnus Rueping, Jorge Gascon, Huabin Zhang
Angew. Chem. Int. Ed. 2025, e202517916
DOI: 10.1002/anie.202517916
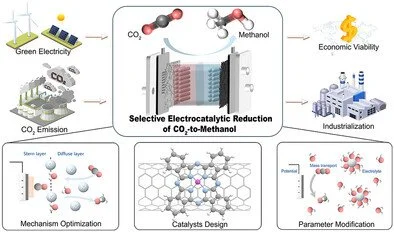
Abstract:
Electrocatalytic CO2 reduction to methanol (MeOH) unites two urgent global needs, carbon recycling and renewable energy storage, into a single, compelling chemical transformation. According to recent techno-economic analyses, commercially competitive MeOH production (at ≈$190 per ton) can be achieved via electroreduction by meeting practical targets for current density, Faradaic efficiency (FE), and stability. Moreover, MeOH's high energy density (16 MJ L−1), substantial hydrogen content (100 g H2 per L), and low storage and transport costs further underscore its strong economic potential. Yet, the complexity of the six-electron–proton transfer (ET–PT) process that governs its formation remains intrinsically complex, with competing pathways threatening selectivity at every stage. This review critically examines current mechanistic insights, highlighting key intermediates such as CO and OCH3, and demonstrating how catalyst surfaces and reaction conditions profoundly influence pathway divergence. We highlight recent advances in catalyst development that exploit a fundamental, molecular-level understanding of intermediate stabilization to deliver unprecedented MeOH selectivity and activity. Through detailed analysis of operational parameters—including mass transport dynamics, electrolyte composition, and applied potentials—this work provides a comprehensive framework for rational catalyst development. Together, these insights converge design principles for next-generation electrocatalysts capable of selectively converting CO2-to-MeOH at scale, advancing economically viable and environmentally sustainable MeOH production.
Moyu Yi, Karthik Peramaiah, Xingzhu Chen, Renqian Zhou, Nursaya Zhumabay, Hongbin Dou, Hao Huang , Indranil Dutta, Shouwei Zuo, Jifeng Wu, Bin Chang, Tsu-Chien Weng, Magnus Rueping, Osman M. Bakr, Huabin Zhang, Kuo-Wei Huang
Applied Catalysis B: Environment and Energy, 2026, 383, 126126
DOI: 10.1016/j.apcatb.2025.126126
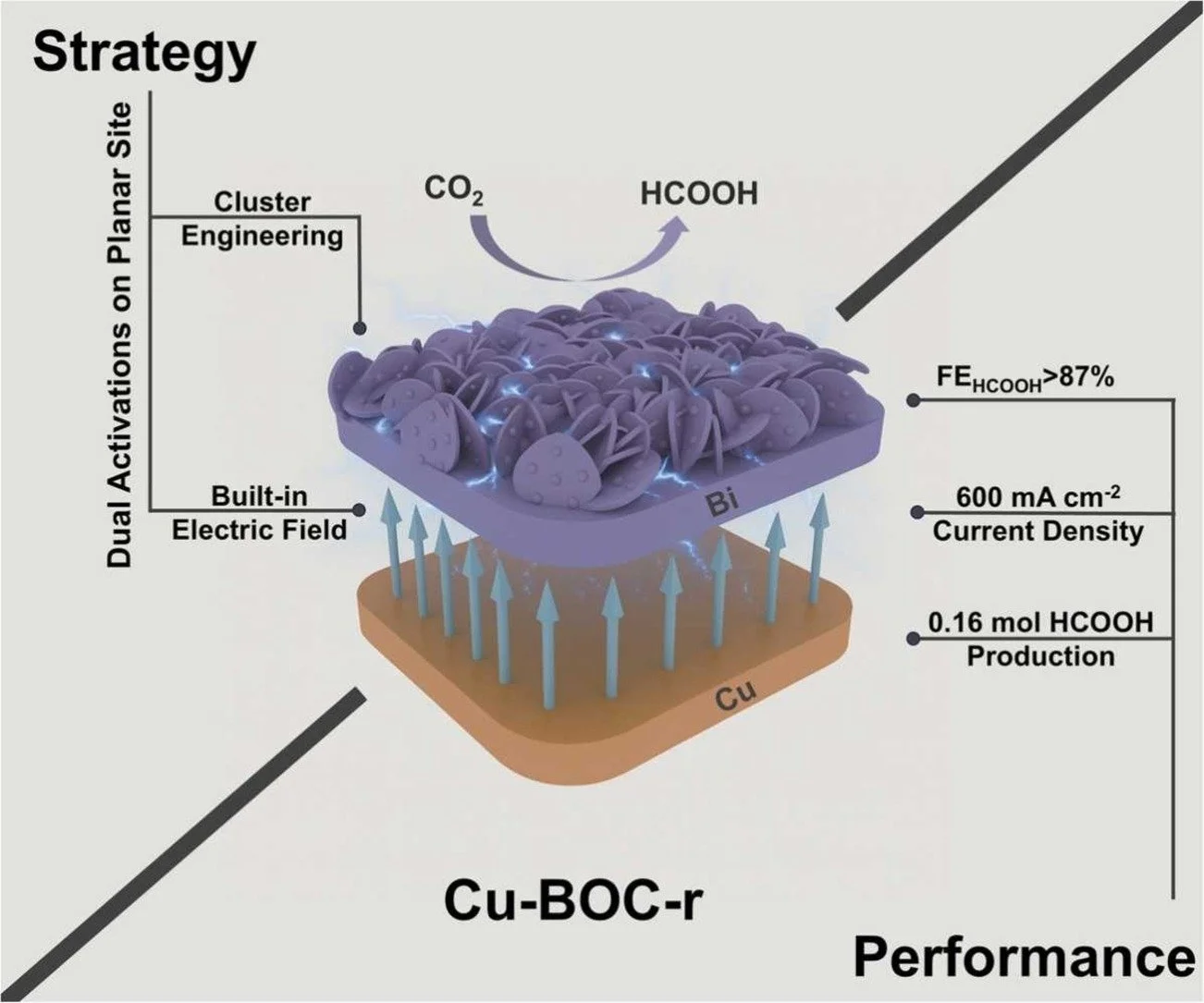
Abstract:
Activating catalytically inert planar sites and achieving high-rate CO2 to formic acid conversion remain key challenges for industrial implementation of bismuth nanoflower-based electrocatalyst. Here, we integrate in-situ reconstruction of Bi nanoclusters with heterointerfacial electric fields at a Bi/Cu junction. The dual (geometric + electronic) activation creates secondary cluster sites, enhances interfacial charge transformation, and improves the selectivity to the key product formic acid. The resulting catalyst achieves > 87 % Faradaic efficiency across a wide current density range (100–600 mA cm−2) and enables mole-scale production of pure formic acid (≈ 0.16 mol) over 100 h of continuous operation in a solid electrolyte reactor. In-situ studies and DFT calculations reveal a synergistic mechanism of planar surface activation by cluster engineering and electric field-enhanced intermediate binding. This work establishes a scalable and integrative catalyst-interface-reactor framework for practical CO2 electrolysis toward a liquid hydrogen carrier.
Titanium Nitride Films as Durable, Enhancing Windows for Electrochemical Infrared Spectroscopy
Nursaya Zhumabay, Jeremy A. Bau, Laurentiu Braic, Huabin Zhang, Yun Hau Ng, Magnus Rueping
ACS Appl. Mater. Interfaces 2025, 17, 61377–61385
DOI: 10.1021/acsami.5c10168
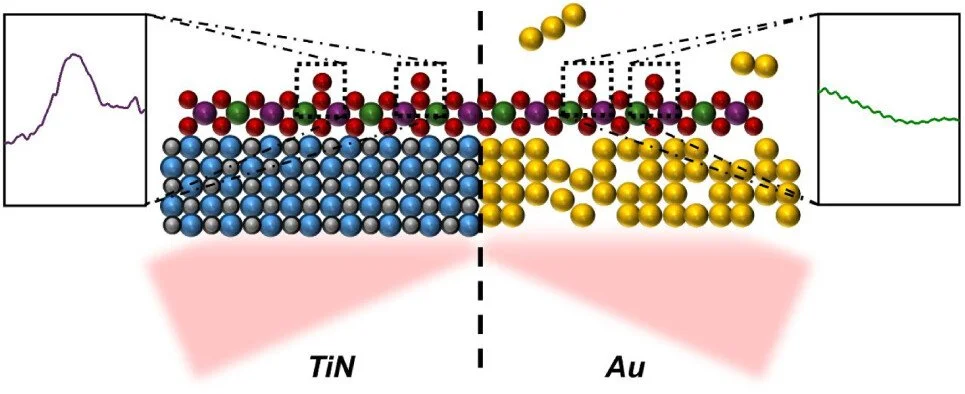
Abstract:
As electrochemical reactions become more central to the green energy transition, spectroscopic means to characterize these reactions are becoming more valuable. Electrochemical attenuated total reflectance – surface enhanced infrared spectroscopy (ATR-SEIRAS) is of particular note for its ability to characterize molecules at the catalyst-electrolyte interface. However, extensive studies using electrochemical ATR-SEIRAS are limited by the poor chemical and mechanical stability or optical properties of window materials. In this work, we report titanium nitride (TiN) prepared in a single-step reactive sputtering process with Ar and N2 plasma as a new material for ATR-SEIRAS on account of its ease of preparation, good conductivity, outstanding mechanical and chemical stability, and ability to acquire surface-enhanced spectra. After depositing Pt on the TiN surface, a CO probe is used to demonstrate the spectroscopic utility of the TiN layers. TiN also shows remarkable chemical stability in the same strong alkaline (1 M KOH) and acidic (0.5 M H2SO4) conditions often used for energy-related studies, especially in comparison to the classic electroless deposited Au system typically utilized. Finally, we validate TiN for studying energy-relevant reactions, demonstrating that meaningful spectra can be collected in CO2 reduction and O2 evolution. These properties make TiN one of the most promising optical window materials yet reported for electrochemical ATR-SEIRAS.
Dr. Liang Yi, Dr. Anton S. Makarov, Dr. Bholanath Maity, Dr. Deshen Kong, Rawan Alshehri, Luigi Cavallo, Rene M. Koenigs, Magnus Rueping
Angew. Chem. Int. Ed. 2025, e18508
DOI: 10.1002/anie.202518508
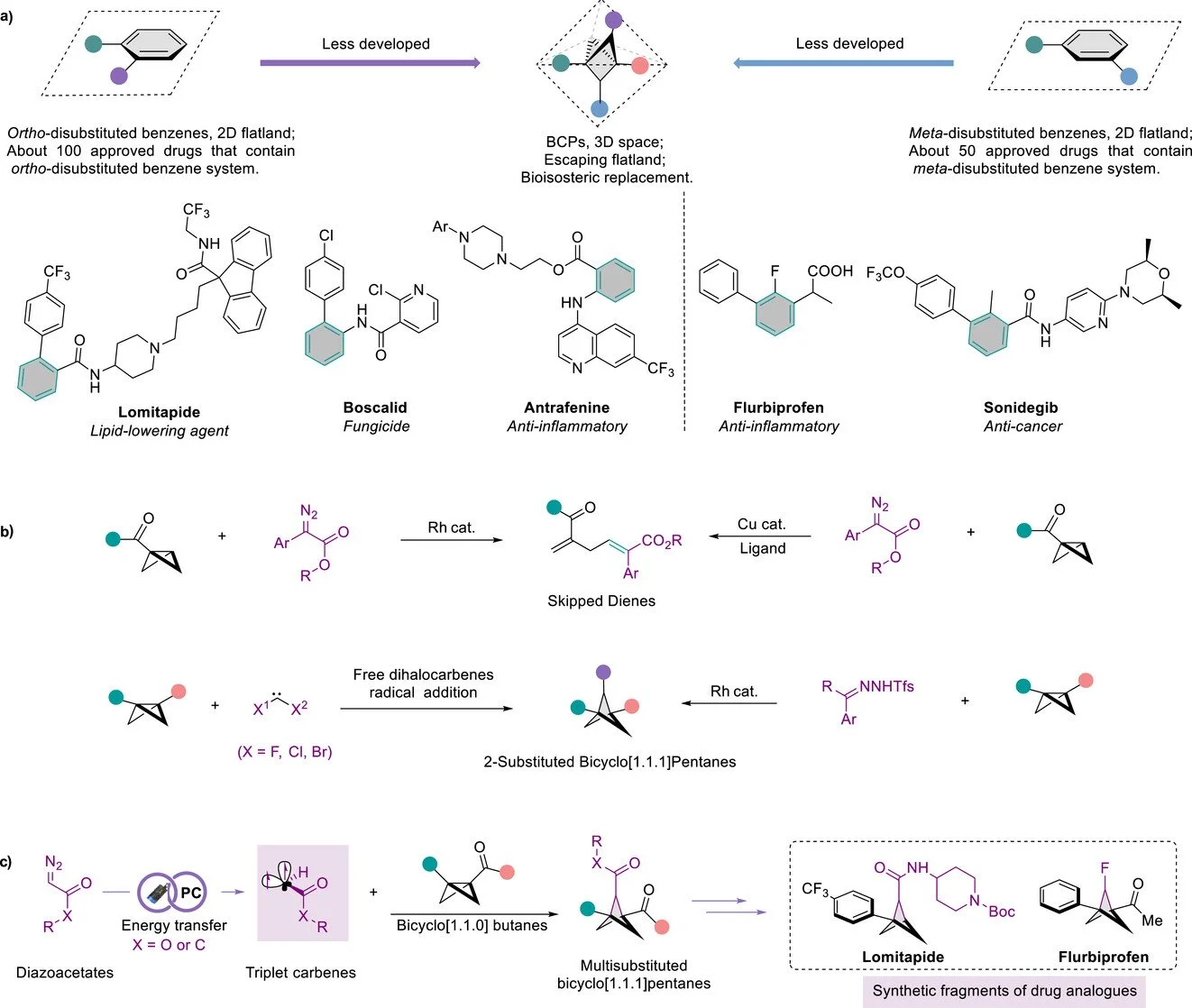
Abstract:
Substituted bicyclo[1.1.1]pentanes (BCPs) are increasingly recognized as valuable sp3-rich bioisosteres for ortho- or meta-disubstituted benzene rings, offering improved pharmacokinetic properties and broad utility in drug discovery. Herein, we report an energy-transfer-enabled strategy for the synthesis of 2-substituted BCPs via strain-release triplet carbene addition to bicyclo[1.1.0]butanes (BCBs). This method employs readily accessible diazoacetates and a diverse set of BCBs under mild blue-light irradiation, providing direct access to 1,2,3-trisubstituted BCPs. Key advantages include the modular installation of versatile bridge ester substituents, enabling downstream diversification, while retaining the bridgehead substituents from the BCB precursor. The synthetic utility is further demonstrated through the preparation of BCP analogues of bioactive fragments. Combined experimental and DFT studies support a mechanism involving photoinduced energy transfer to generate a triplet carbene, followed by stepwise addition across the highly strained C─C σ bond of the BCB framework.
Nanobodies for NETosis Tracking and Visualization
Nicoleta Gutu, Yuli Peng, Dominik Renn, Magnus Rueping
Current Protocols, 2025, 5 , e70213
DOI: 10.1002/cpz1.70213
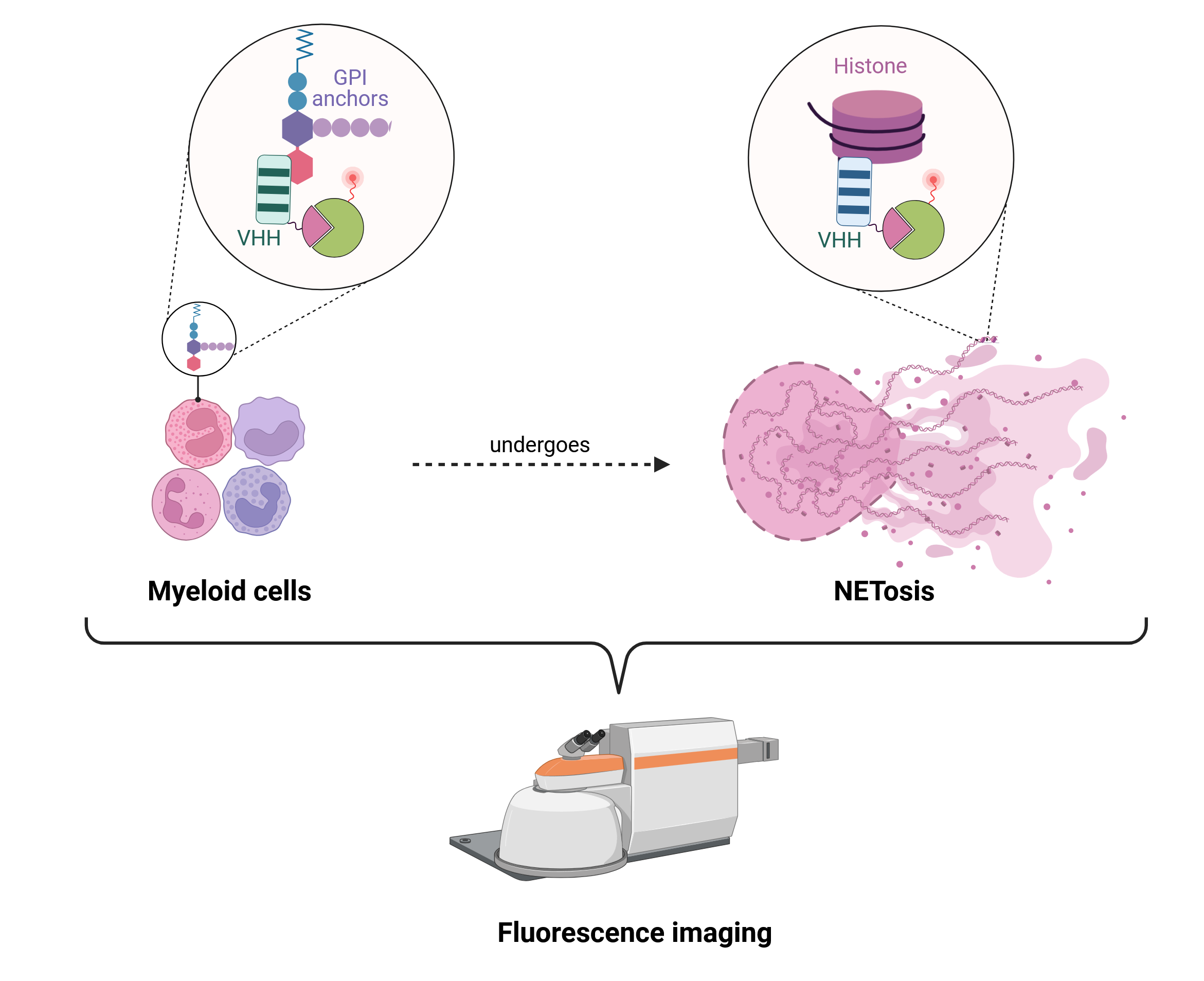
Abstract:
Neutrophil extracellular traps (NETs) are pivotal in the immune response, trapping and neutralizing pathogens. The process of NET formation, or NETosis, is a critical innate immune response mechanism implicated in diverse physiological and pathological processes, including infections, chronic inflammation, and cancer. Accurate imaging of NETosis is essential for unraveling its mechanisms and therapeutic implications, although limitations of specificity and flexibility remain. In this article, we present modified nanobodies targeting Ly6C/G and histones for imaging of NETosis. Single-domain antibodies (VHHs), also known as nanobodies, targeting Ly6C/G specifically bind to neutrophils, and anti-histone nanobodies selectively target NET-associated histones, enabling NETosis monitoring. These nanobodies are engineered to optimize fluorescent probe labeling and can enable high-resolution visualization of NETosis through whole-body fluorescence tomography and/or intravital microscopy. Our approach offers an alternative for expanding the existing NETosis imaging toolkit.
Basic Protocol 1: Cloning, cytoplasmic expression, and purification of proteins
Basic Protocol 2: Fluorophore binding assay
Support Protocol 1: Analysis by gel staining and immunoblotting
Support Protocol 2: In-gel tryptic digestion and sample preparation for LC-MS/MS analysis
Support Protocol 3: SpyCatcher003/SpyTag003 binding assay
Swati Jindal, Kuntal Pal, Mostafa Zeama, Partha Maity, Tian Jin, Mickaele Bonneau, Rajesh Kancherla, Omar F. Mohammed, Osama Shekhah, Magnus Rueping, Mohamed Eddaoudi
Chem., 2025, 12, 102751
DOI: 10.1016/j.chempr.2025.102751
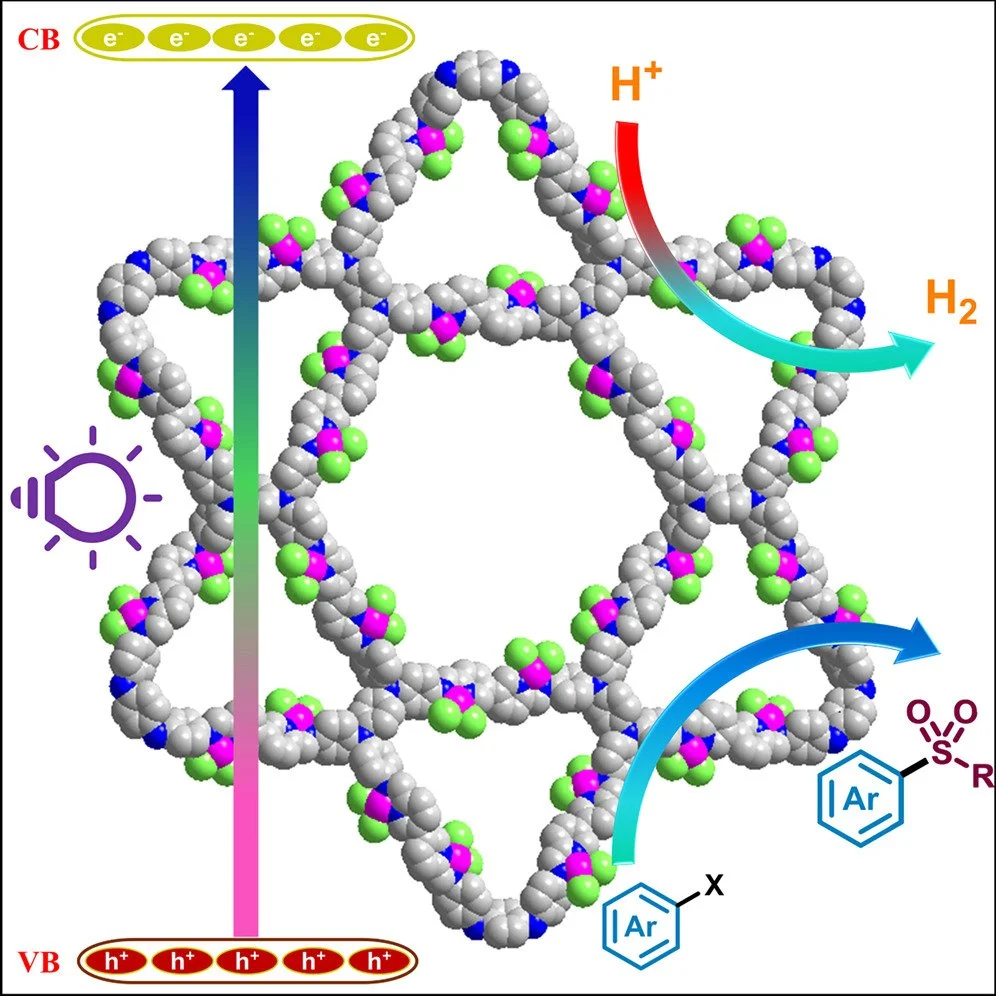
Abstract:
This work reports the design and synthesis of a novel imine-linked 2D covalent organic framework (COF), TPDA-BiPy-COF, constructed from [3,3′ -bipyridine]-6,6′ -dicarboxaldehyde (3,3′ -BiPy) as an electron acceptor and tetrakis(4-aminophenyl)-1,4-phenylenediamine (TPDA) as a donor. The COF features pyridylimine linkages, i.e., Nimine-Ni-Nbipyridine, with active nitrogen sites that facilitate proton reduction to hydrogen. To improve photocatalytic hydrogen evolution performance, Ni(II) centers were introduced via post-synthetic metalation, forming TPDA-BiPy@NiX₂ COF (X = Cl, Br). The coordination of Ni(II) with the imine and bipyridine nitrogen atoms enhanced framework planarity and conjugation, thereby boosting photocatalytic activity. Notably, TPDA-BiPy@Ni(II) COF achieved an excellent hydrogen evolution rate of 34.13 mmol g⁻¹ h⁻¹ under visible light, without requiring a cocatalyst. Furthermore, the metallaphotoredox activity of TPDA-BiPy@Ni (II) displayed its promise for photocatalyzed C–S cross-coupling reaction. This dual-functional catalyst highlights the advantage of incorporating nickel into COFs, offering a cost-effective and sustainable alternative to noble-metal-based systems for photocatalysis and synthetic transformations.
Bridging Scales in Solar-Driven Water Splitting: Pathways to System Integration
Chengyang Feng, Miao Hu, Jumanah Alharbi, Magnus Rueping, Huabin Zhang
Adv. Mater., 2025, 37, e06690
DOI: 10.1002/adma.202506690
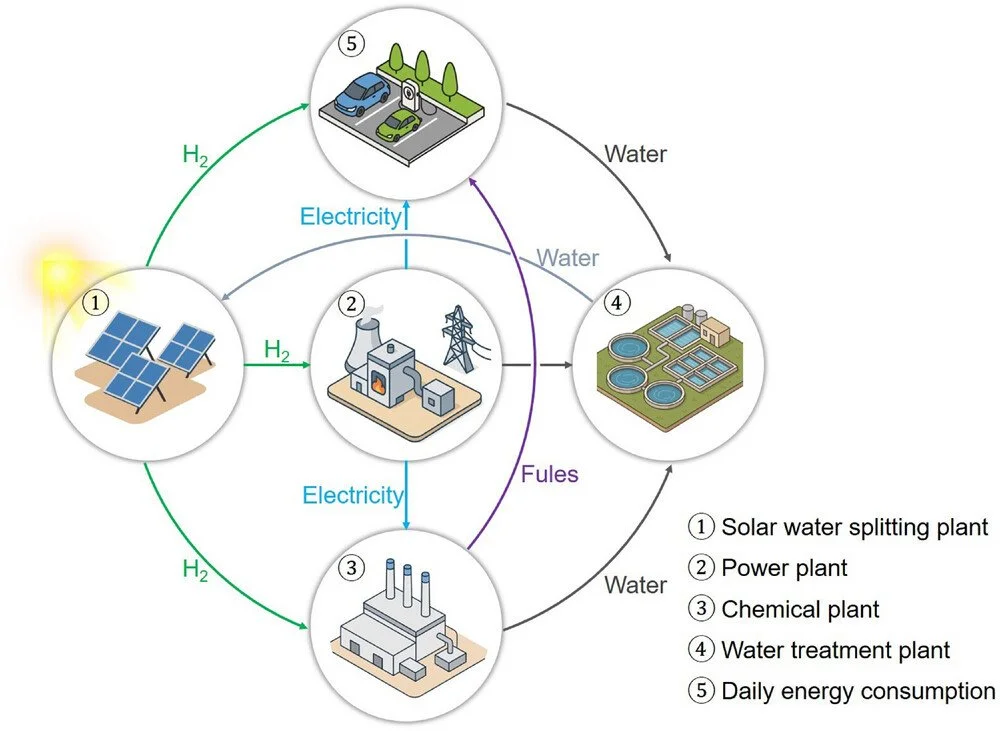
Abstract:
Artificial photosynthesis, which converts and stores solar energy as chemical energy, holds immense potential for promoting sustainable development and achieving carbon neutrality. Solar-driven water splitting offers an ideal method for storing solar energy, with one of the most promising approaches based on efficient particulate photocatalysts. In recent years, significant progress has been made in particulate photocatalyst-based water splitting systems, from fundamental scientific research to exploratory practical applications. However, to date, no photocatalytic water splitting system has achieved the efficiency required for practical applications. The development of high-performance photocatalysts and optimized photocatalytic systems is urgently needed. This review examines the crucial factors limiting the activity of photocatalysts for overall water splitting and summarizes design strategies to enhance photocatalyst performance and overcome these barriers. The design and modification strategies for high-efficiency photocatalysts are highlighted, including bandgap regulation, localized surface plasmon resonance, morphology control, crystal facet engineering, heterostructures, cocatalysts, and external-field association. Additionally, the scalability of using particulate photocatalysts for overall water splitting driven by natural sunlight is discussed. Finally, insights into advanced strategies for improving the performance of particulate photocatalysts are provided, and perspectives on the future development of solar water splitting systems for commercial applications are offered.
Prashant S. Shinde, Valmik S. Shinde, Chen Zhu, Magnus Rueping
ACS Catal. 2025, 15, 17198–17205
DOI: 10.1021/acscatal.5c05140
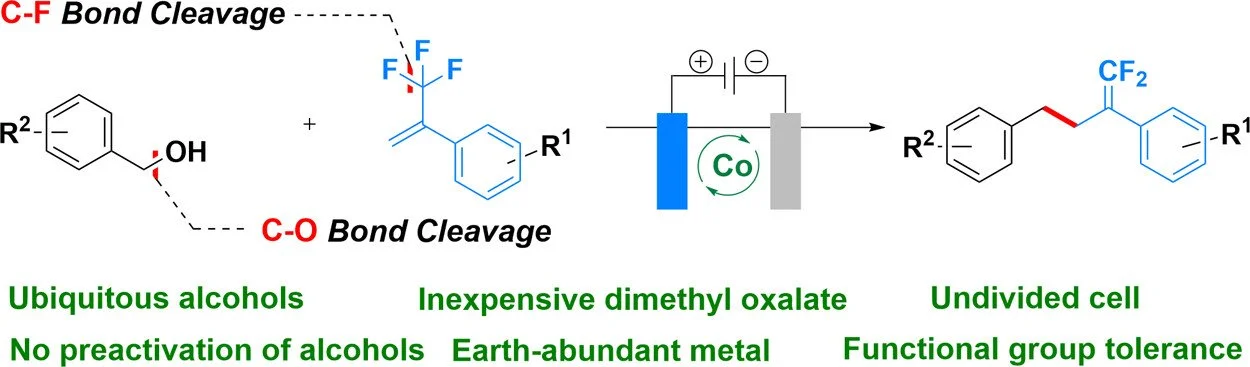
Abstract:
We report an efficient cross-electrophile coupling strategy for the synthesis of gem-difluoroalkenes from readily available benzyl alcohols and α-trifluoromethyl alkenes. This deoxygenative/defluorinative transformation proceeds via the simultaneous cleavage of strong C–O and C–F bonds under mild electrochemical conditions, generating highly reactive benzyl radicals in situ. The methodology eliminates the need for multistep alcohol preactivation protocols and exhibits a broad substrate scope with functional group tolerance. Its synthetic utility is further demonstrated by gram-scale reactions and the late-stage functionalization of complex molecules. Given the ubiquity of alcohols and the importance of gem-difluoroalkene motifs in pharmaceutical and materials chemistry, this protocol offers a practical and scalable platform for C–C bond formation via radical cross-electrophile coupling.
Iron-Catalyzed Reductive Alkylation of Imines via Ligand-to-Metal Charge Transfer
Serik Zhumagazy, Kuntal Pal, Magnus Rueping
ChemCatChem 2025, 17, e01210
DOI: 10.1002/cctc.202501210
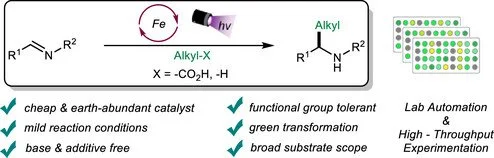
Abstract:
Herein, we report a sustainable, iron-catalyzed reductive alkylation reaction of imines with carboxylic acids and unactivated hydrocarbons to access synthetically relevant amine motifs. The reaction proceeds via a light-induced ligand-to-metal charge transfer (LMCT) process, generating alkyl radicals either through a decarboxylative pathway or by forming a reactive chlorine radical that facilitates hydrogen atom transfer (HAT) with alkanes. This photochemical transformation offers several advantages, including sustainability, mild reaction conditions, cost efficiency, and an excellent atom economy.
Yang Li, Zhen Cao, Cailing Chen, Guoxiang Zhao, Edy Abou-Hamad, Zhi-Peng Wu, Zirui Wang, Qiaohong Li, Magnus Rueping, Yu Han, Luigi Cavallo, Huabin Zhang
J. Am. Chem. Soc., 2025, 147, 34933–34943
DOI: 10.1021/jacs.5c11417
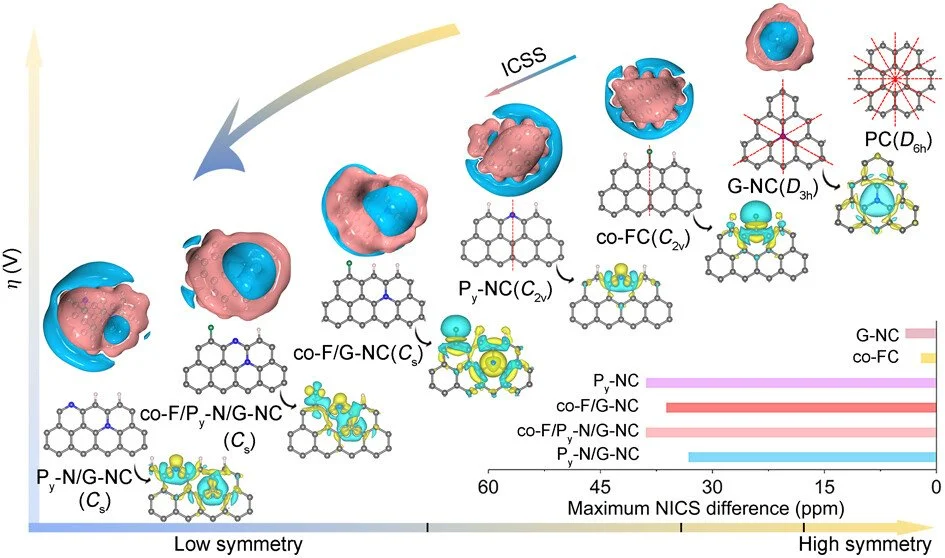
Abstract:
The atom arrangement in carbon electrocatalysts is crucial for enhancing the intrinsic activity toward oxygen reduction reactions (ORRs), a key process in multiple renewable energy systems. However, the challenge of designing electrocatalysts with improved performance by manipulating atomic arrangement has been limited by synthetic constraints and a lack of understanding of the catalytic phase formation. Herein, we gain atomic-level insight into the origin of a highly active site by creating a model catalyst with a heteroatom-decorated carbon matrix of a specific configuration. The introduction of fluorine (F) during the synthesis of the nitrogen (N)-decorated carbon matrix induces structural rearrangement, converting most pyrrolic-N (Pr-N) into highly stable graphitic-N (G-N), thereby achieving a N configuration predominantly composed of pyridinic nitrogen (Py-N) and G-N. The multidopant synergistic effect of F, Py-N, and G-N causes a destabilized π-conjugated electron network of the carbon matrix, resulting in a more localized electronic structure. As a result, multiple dopant configurations with high ORR activity have been explored, among which the asymmetric Py-N and G-N configurations feature the lowest theoretical ORR overpotential, ultimately enabling the optimized F@NC catalyst to exhibit excellent oxygen reduction activity. This work establishes a foundation for the rational design of metal-free carbon-based electrocatalysts toward ORR.
Nanodesigner: resolving the complex-CDR interdependency with iterative refinement
Melissa Maria Rios Zertuche, Şenay Kafkas, Dominik Renn, Magnus Rueping, Robert Hoehndorf
J Cheminform, 2025, 17, 120
DOI: 10.1186/s13321-025-01069-2
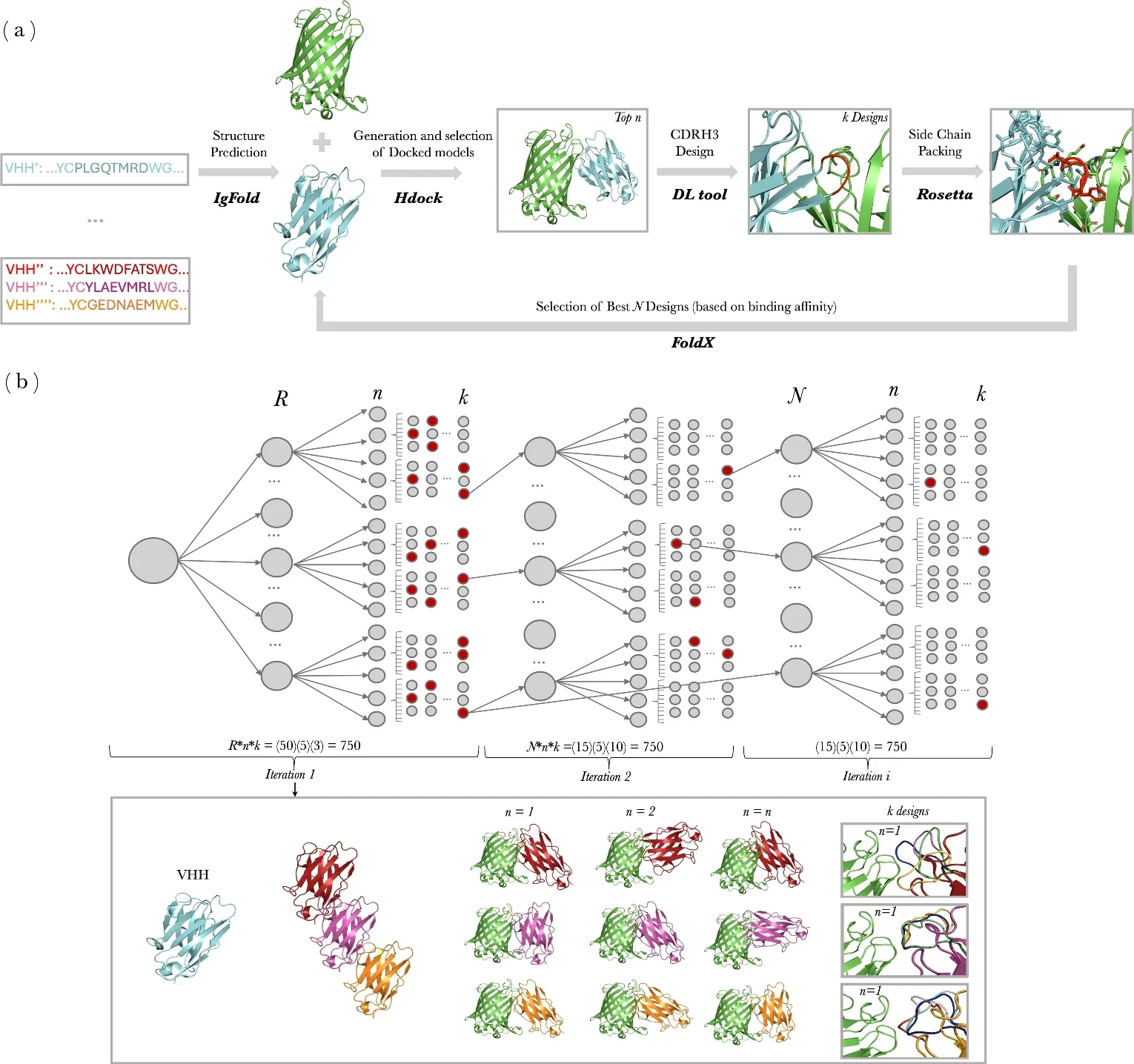
Abstract:
Camelid heavy-chain only antibodies consist of two heavy chains and single variable domains (VHHs), which retain antigen-binding functionality even when isolated. The term “nanobody” is now more generally used for describing small, single-domain antibodies. Several antibody generative models have been developed for the sequence and structure co-design of the complementarity-determining regions (CDRs) based on the binding interface with a target antigen. However, these models are not tailored for nanobodies and are often constrained by their reliance on experimentally determined antigen–antibody structures, which are labor-intensive to obtain. Here, we introduce NanoDesigner, a tool for nanobody design and optimization based on generative AI methods. NanoDesigner integrates key stages—structure prediction, docking, CDR generation, and side-chain packing—into an iterative framework based on an expectation maximization (EM) algorithm. The algorithm effectively tackles an interdependency challenge where accurate docking presupposes a priori knowledge of the CDR conformation, while effective CDR generation relies on accurate docking outputs to guide its design. NanoDesigner approximately doubles the success rate of de novo nanobody designs through continuous refinement of docking and CDR generation.
Mohammed Faihan Alotaibi, Arunachalam Sagadevan, Peng Yuan, Mohammad Bodiuzzaman, Kathiravan Murugesan, Renqian Zhou, Jun Yin, Simil Thomas, Ren-Wu Huang, Naveen M. Halappa, Chunwei Dong, Mutalifu Abulikemu, Husam N. Alshareef, Omar F. Mohammed, Magnus Rueping, Osman M. Bakr
J. Am. Chem. Soc., 2025, 147, 28932-28942
DOI: 10.1021/jacs.5c06846
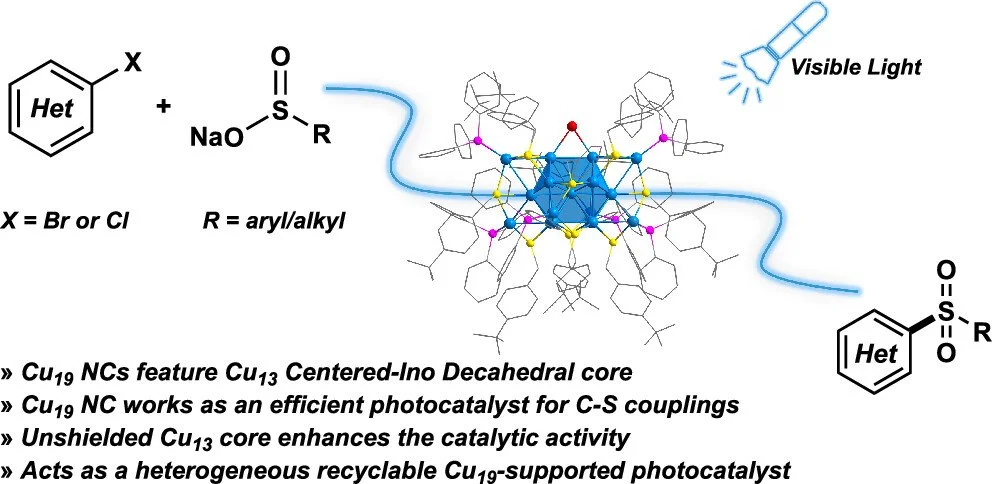
Abstract:
The synthesis of metal nanoclusters (NCs) having decahedral cores is challenging, with only a few reports primarily focusing on systems containing gold (Au) and silver (Ag). Herein, we present the synthesis, characterization, and catalytic activities of a copper (Cu) NC featuring a centered Ino decahedral core: Cu19(tBuPhCH2S)12(PPh3)6H6Br (Cu19). Unlike typical core–shell structured NCs, Cu19 exhibits a unique structure with a core capped by four motifs from four directions, where two square faces and both apexes of the Ino decahedron core are capped, leaving three square faces of the core exposed, acting as highly efficient catalytic active sites. This distinctive design opens new possibilities for catalytic applications of Cu NCs, as demonstrated experimentally in the sulfonylation of aryl halides under visible light irradiation at room temperature. Density functional theory (DFT) and experimental results suggest that the catalytic cycle proceeds through a single-electron transfer (SET) process between the photoexcited Cu19 NC-sulfinates and the aryl halide, enabling efficient C–S arylation of sulfinates. The unshielded Cu19 NC catalysts can be converted into a heterogeneous recyclable catalyst and are compatible with a wide range of aryl halides and sulfinate nucleophiles for producing sulfones.
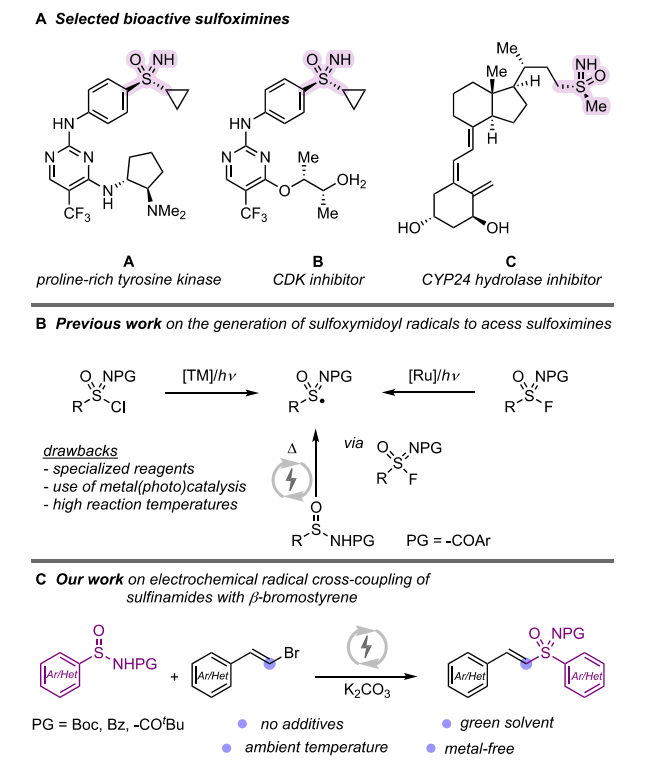
Electrochemical S-vinylation of sulfinamides with β-bromostyrenes
Yaseen Hussain, Ivan Sliusarevskyi, Claire Empel, Magnus Rueping, Rene M. Koenigs
Chem. Commun., 2025, 61, 11017-11020
DOI: 10.1039/D5CC02045J
Abstract:
We present an electrochemical method for sulfoximine synthesis via radical cross-coupling of sulfonimidoyl and styryl radicals, generated from sulfinamides and bromostyrenes. This approach enables the efficient synthesis of vinyl sulfoximines, including bioactive-tethered derivatives, in moderate to good yields.
Small organic fluorophores with SWIR emission detectable beyond 1300 nm
Michal Pieczykolan, Pierre Alix Dancer, Tjadina-Wencke Klein, Hubert Piwonski, Hannes Rolbieski, Bholanath Maity, Oliver T. Bruns, Luigi Cavallo, Fabian Kiessling, Magnus Rueping, Srinivas Banala
Chem. Commun., 2025, 61, 4820-4823
DOI: 10.1039/D4CC05248J

Abstract:
3,6-Dimethylamino fluorenone was functionalized with substituents to achieve an absorption maximum at 1012 nm and emission >1300 nm. TD-DFT calculations confirmed that the substituent orbitals contribute to narrowing the HOMO–LUMO energy gap. Imaging with an InGaAs-based SWIR camera and various longpass filters confirmed detection >1300 nm.
Sharath Kandambeth, Rajesh Kancherla, Kuntal Pal, Taslim Melliti, Mostafa Zeama, Vinayak S. Kale, Issatay Nadinov, Abdulaziz M. Alali, Osama Shekhah, Omar F. Mohammed, Magnus Rueping, Mohamed Eddaoudi
Angew. Chem. Int. Ed. 2025, e202503328
DOI: 10.1002/anie.202503328
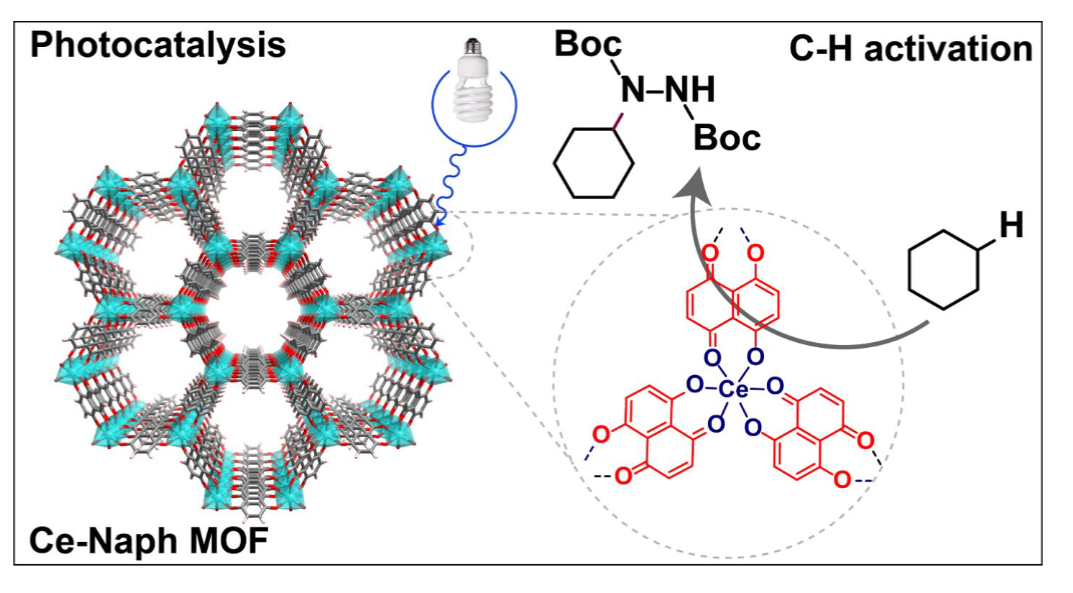
Abstract:
In this work we have successfully synthesized a series of novel semiconducting 2D catecholate metal-organic frameworks (MOFs) based on naphthazarin ligands by utilizing unraveled metal-acetyl acetonate linkage chemistry. The synthesized Ce-Naph MOF exhibited excellent light absorption properties and chemical stability across various solvents. Its insoluble and stable framework, combined with an optimal band gap, enabled its use as a photocatalyst for organic transformations. For the first time, Ce-Naph MOF is explored as a heterogeneous catalyst for photocatalytic applications specifically for the selective C–H amination of alkanes, achieving yields of up to 89% under ambient conditions. We propose that the initial metal-to-ligand charge transfer in Ce-Naph MOF, promoted by light, is essential for forming an active alkoxy-Ce(IV)-species. This species subsequently undergoes ligand-to-metal charge transfer to generate the alkoxy radical, which acts as a hydrogen atom transfer reagent to activate alkanes. Furthermore, Ce-Naph MOF demonstrated long-term cyclic stability, maintaining its catalytic activity and structural integrity over five cycles, highlighting its durability as a heterogeneous catalyst. We are confident that this straightforward and practical methodology opens new avenues for industrial applications, significantly advancing the fields of metal catalysis, photocatalysis, and sustainable chemistry.
Selective Alkylarylation Difunctionalization of 1,3-Butadienes via Nickel/Photoredox Dual Catalysis
Aidana Gimnkhan, Rajesh Kancherla, Krishnamoorthy Muralirajan, Magnus Rueping
Org. Lett. 2025, 27, 5509-5514
DOI: 10.1021/acs.orglett.5c01518

Abstract:
Photocatalytic multicomponent reactions are at the forefront of organic synthesis. In recent years, the dicarbofunctionalization of olefins has received significant attention, with nickel serving as a key transition metal catalyst under photochemical conditions. However, achieving regioselective 1,4-alkylarylation of dienes with alkyl and aryl bromides remains challenging. In this work, we present a Ni/photoredox dual catalysis approach for the regioselective alkylarylation of dienes, offering a mild reaction that eliminates the need for stochiometric metal reductants. Broad substrate scope and mechanistic investigations are presented that support the proposed reaction mechanism.
Palladium-Catalyzed Alkyl Amination of Olefins via Radical-Polar Crossover at Room Temperature
Anurag Singh, Kuntal Pal, Sayan Dutta, Arnab Dey, Rajesh Kancherla, Bholanath Maity, Luigi Cavallo, Magnus Rueping
Angew. Chem. Int. Ed. 2025, e202503446
DOI: 10.1002/anie.202503446

Abstract:
In contrast to traditional ground-state palladium catalyzed alkyl Heck reactions, which are thermodynamically unfavourable and endothermic, excited-state palladium catalysis facilitates single-electron mechanisms, with light primarily driving the formation of alkyl radicals from triplet-state Pd(0). Here, we report a novel and mechanistically distinct Pd-catalyzed reaction, where the key hybrid alkyl Pd(I)-radical intermediate is generated from the triplet-state Pd(II) at room temperature. This hybrid species engages in the addition to dienes and conjugated enynes, producing a transposed open-shell allyl Pd(I)-radical, which undergoes radical-polar crossover (RPC) to yield the desired alkyl amination products. Density functional theory (DFT) studies offer insights into the reaction mechanism, confirming the involvement of hybrid alkyl/allyl Pd(I) radical species as key intermediates.
A formal vinylogous Schmidt reaction: nitrogen insertion of para-quinone methides
Bo Wang, Chen Zhu, Yanhao Dong, Chen Kong, Hanxiao Zhu, Tiandi Ding, Guoyue Wei, Yatong Yang, Xin Zhao, Magnus Rueping, Qingqiang Yao, Kun Zhao, Yan Li, Ying Zhi
Org. Chem. Front., 2025, 12, 5245-5251
DOI: 10.1039/D5QO00476D

Abstract:
Compared with recent nitrogen addition reactions, which mainly focus on the styrene motif, feasible modification of a conjugated C(sp2)–C(sp2) single bond is rare due to its robustness. Herein, we report a facile nitrogen introduction to the conjugated C(sp2)–C(sp2) bond of p-QMs through a vinylogous Schmidt process, using N-OTs carbamate as a bench-stable ambiphilic nitrogen source. Depending on the electron-donating ability of the ortho-substituent on the benzene ring, the reaction underwent the formal vinylogous Schmidt process through an aziridine intermediate or a 1,6-addition/cyclization step delivering benzoxazolidine or dihydroindazole scaffolds, respectively. This study not only expands the application boundary of the Schmidt reaction but also provides a new strategy for nitrogen addition to a C(sp2)–C(sp2) bond.
Nanobody-Based Lateral Flow Assay for Rapid Zika Virus Detection
Yuli Peng, Atheer Alqatari, Fabian Kiessling, Dominik Renn, Raik Grünberg, Stefan T. Arold, Magnus Rueping
ACS Synth. Biol. 2025, 14, 890–900
DOI: 10.1021/acssynbio.4c00819

Abstract:
Zika virus infections remain severely underdiagnosed due to their initial mild clinical symptoms. However, recent outbreaks have revealed neurological complications in adults and severe deformities in newborns, emphasizing the critical need for accurate diagnosis. Lateral flow assays (LFAs) provide a rapid, cost-effective, and user-friendly method for antigen testing at point-of-care, bedside, or in home settings. LFAs utilizing nanobodies have multiple benefits over traditional antibody-based techniques, as nanobodies are much smaller, more stable, and simpler to manufacture. We introduce a nanobody-based LFA for the rapid identification of Zika virus antigens. Starting from two previously reported nanobodies recognizing the Zika nonstructural protein 1 (NS1), we evaluate periplasmic and cytosolic nanobody expression and test different purification tags and immobilization strategies. We quantify nanobody binding kinetics and validate their mutually noncompetitive binding. Avidity effects boost the capture of the tetrameric target protein by 3 orders of magnitude and point to a general strategy for higher sensitivity LFA sensing. The nanobody LFA detects Zika NS1 with a limit of detection ranging from 25 ng/mL in buffer to 1 ng/mL in urine. This nanobody-LFA has the potential to facilitate on-site and self-diagnosis, improve our understanding of Zika infection prevalence, and support public health initiatives in regions affected by Zika virus outbreaks.
Electrochemical C-H functionalization reaction of N-heterocycles with alkyl iodides
Yaseen Hussain, Ganga Sankar, Magnus Rueping Rene M. Koenigs
Chem. Commun., 2025, 61, 8691-8694
DOI: 10.1039/D5CC01836F

Abstract:
Herein we report an electro catalyzed alkylation of heterocycles with alkyl halide through a XAT strategy. A wide range of alkyl halides (Br, I) were tolerated to deliver the alkylated heterocycles. Further, azauracil bearing various functionalities and bioactive molecules viz. isozepac, borneol, menthol etc. also delivered the corresponding alkylated products in acceptable yield. We consider that the reaction proceeds by initial oxidation of triethylamine under electrochemical conditions, followed by deprotonation to α-amino radical intermediate. Then the radical intermediate abstracts the halogen atom from the alkyl halide to generate the alkyl radical and α-halo amine. Finally, the intermediate adds onto the imine carbon followed by deprotonation and subsequent oxidation to provide the esired product.
Deshen Kong, Liang Yi, Alice Nanni, Magnus Rueping
Nat Commun, 2025, 16, 3983
DOI: 10.1038/s41467-025-59358-1

Abstract:
Photocatalysis has greatly advanced in organic synthesis but still confronts challenges, including light attenuation in reaction media and excessive solvent utilization. These issues lead to inefficiencies, particularly in heterogeneous cloudy mixtures and in scaling-up applications. Integrating photocatalysis with mechanochemistry offers a nascent but promising solution to these challenges. Herein, we present a scalable photo-mechanochemical platform that combines visible-light photocatalysis with Resonant Acoustic Mixing (RAM), enabling efficient cross-coupling reactions under solvent-minimised conditions. This approach demonstrates broad substrate tolerance, accommodating a variety of aryl (hetero) halides and N-, O-, P-, S-nucleophiles. The protocol supports scaling up to 300 mmol, representing a 1500-fold increase, while maintaining exceptionally low catalyst loading and achieving up to 9800 turnover numbers (TON). The generality of this platform is further validated by its applicability to other synthetic transformations.
Gentoku Takasao, Bholanath Maity, Sayan Dutta, Rajesh Kancherla, Magnus Rueping, Luigi Cavallo
ACS Catal. 2025, 15, 5915−5927
DOI: 10.1021/acscatal.4c06474

Abstract:
We present an in silico workflow to streamline the identification of promising N-heterocyclic carbenes (NHCs) as ligands in metal catalysis or as catalysts in organocatalysis. Central to this workflow is the NHC-cracker database, which contains over 200 descriptors for 1781 nonredundant NHCs, each documented as an NHC-metal complex in the Cambridge Structural Database. To demonstrate its utility, we applied it to two catalytic problems using literature data. First, we analyzed 21 Ru–NHC complexes active in the ethenolysis of cyclic olefins. An MLR (multivariate linear regression) model trained on 11 Ru complexes based on NHCs in NHC-cracker successfully rationalized the behavior of the remaining 10 complexes. Second, we examined an Ir–Ni dual-catalyzed Csp2–Csp3 cross-coupling reaction involving five experimentally tested NHC skeletons. Using a multiscale workflow, we created DFT-based data sets to train two MLR models: one for productive substrate activation and another for detrimental NHC dimerization. Consistent with experiments, the models identified oxazoles as reactive, while benzimidazoles, triazoles, thiazoles, and untested cyclic (alkyl)(amino)carbenes were predicted as nonreactive. Experimental validation confirmed the latter’s lack of productive substrate activation, supporting the proposed mechanistic scenario.
Prashant S. Shinde, Valmik S. Shinde, Magnus Rueping
Chem. Sci., 2025, 16, 6273-6281
DOI: 10.1039/D5SC00297D

Abstract:
We report a Ni-catalyzed cascade reaction leading to the arylation of an alkyne-induced acyl migration and the formation of all-carbon tetra-substituted alkenes in good yields with exclusive Z-selectivity. This transformation involves the generation of a nucleophilic vinyl-Ni species through regioselective syn-aryl nickelation of the alkynes, followed by an intramolecular acyl migration. The steric and electronic properties of the phosphine ligands are crucial for achieving high regio- and stereocontrol in this migratory carbo-acylation process. The synthetic utility of the resulting Z-tetra-substituted alkenes is also demonstrated.
Ru-OV Site-Mediated Product Selectivity Switch for Overall Photocatalytic CO2 Reduction
C. Feng, M. Hu, S. Zuo, J. Luo, P. Castaño, Y. Ren, M. Rueping, H. Zhang
Adv. Mater. 2025, 37, 2411813
DOI:10.1002/adma.202411813

Abstract:
The photocatalytic reduction of carbon dioxide (CO2) to methane (CH4) represents a sustainable route for directly converting greenhouse gases into chemicals but poses a significant challenge in achieving high selectivity due to thermodynamic and kinetic limitations during the reaction process. This work establishes Ru-OV active sites on the surface of TiO2 by anchoring coordination unsaturated Ru single-atoms, which stabilize crucial reaction intermediates and facilitate local mass transfer to achieve dual optimization of the thermodynamics and kinetics of the overall photocatalytic CO2 reduction. Combining operando spectroscopy with density functional theory (DFT) calculations indicates that oxygen vacancies (OV) inhibits the desorption of *CO, whereas Ru facilitates proton extraction. This configuration not only lowers the overall activation energy barrier but has also been engineered to serve as a selectivity switch, changing the reaction route to produce CH4 instead of CO. Consequently, the Ru-OV/TiO2 exhibits a 195.4-fold improvement in the CH4 yield compared to TiO2, accompanied by an increase in selectivity to 81%.
Haifeng Chen, Magnus Rueping
Chem. Sci., 2025, 16, 6317-6324
DOI: 10.1039/D4SC07923J

Abstract:
Electrochemically driven carbon–carbon formation is receiving considerable interest in organic synthesis. In this study, we present an electrochemically driven method for the formation of C(sp3)–C(sp3) bonds using readily available allylic carbonates, as well as primary, secondary, and tertiary alkyl bromides as electrophiles. This approach offers a highly selective route for synthesizing a broad range of allylic products with excellent functional group tolerance, all without the need for transition metal catalysts. Remarkably, this method also enables the smooth late-stage functionalization of various natural product- and drug-derived substrates, yielding the corresponding complex allylalkanes.
Wenting Feng, Bin Chang, Yuanfu Ren, Debin Kong, Hua Bing Tao, Linjie Zhi, Mohd Adnan Khan, Rashed Aleisa, Magnus Rueping, Huabin Zhang
Adv. Mater. 2025, 37, 2416012
DOI: 10.1002/adma.202416012

Abstract:
Proton exchange membrane water electrolysis (PEMWE) represents a promising technology for renewable hydrogen production. However, the large-scale commercialization of PEMWE faces challenges due to the need for acid oxygen evolution reaction (OER) catalysts with long-term stability and corrosion-resistant membrane electrode assemblies (MEA). This review thoroughly examines the deactivation mechanisms of acidic OER and crucial factors affecting assembly instability in complex reaction environments, including catalyst degradation, dynamic behavior at the MEA triple-phase boundary, and equipment failures. Targeted solutions are proposed, including catalyst improvements, optimized MEA designs, and operational strategies. Finally, the review highlights perspectives on strict activity/stability evaluation standards, in situ/operando characteristics, and practical electrolyzer optimization. These insights emphasize the interrelationship between catalysts, MEAs, activity, and stability, offering new guidance for accelerating the commercialization of PEMWE catalysts and systems.
Yuli Peng, Yaning Huang, Fabian Kiessling, Dominik Renn, Magnus Rueping
ACS Synth. Biol. 2025, 14, 420-430
DOI: 10.1021/acssynbio.4c00592

Abstract:
The COVID-19 pandemic has highlighted the critical need for pathogen detection methods that offer both low detection limits and rapid results. Despite advancements in simplifying and enhancing nucleic acid amplification techniques, immunochemical methods remain the preferred choice for mass testing. These methods eliminate the need for specialized laboratories and highly skilled personnel, making home testing feasible. The here developed low-cost, easily producible and nanobody-based LFAs for the rapid detection of SARS-CoV-2 and MERS-CoV adhere to the essential criteria of the World Health Organization’s recommended ASSURED guidelines. The overall design enables rapid adaptation to emerging and evolving virus variants, with the flexibility to incorporate either specifically binding nanobodies or broadly binding nanobodies as needed. Moreover, the results are noticeable to the naked eye, which is important for point-of-care diagnostic. Additionally, this is the first reported nanobody-based MERS-CoV LFA and the first nanobody-based sandwich LFA that can be observed with the naked eye. We established a reliable protocol for conjugating gold nanoparticles with carboxyl groups to nanobodies produced in E. coli. We demonstrate nanobody-based Lateral Flow Assays (LFAs) for the rapid detection of SARS-CoV-2 and MERS-CoV in single and dual formats as point-of-care diagnostic tools. The developed LFAs are highly sensitive and successfully detected analytes at clinically relevant diagnostic cut-off values. Additionally, our results confirmed that the LFAs have a long shelf life and can be produced cost-effectively and with ease.
Ultrafast Charge Transfer on Ru-Cu Atomic Units for Enhanced Photocatalytic H2O2 Production
C. Feng, J. Alharbi, M. Hu, S. Zuo, J. Luo, H. S. Al Qahtani, M. Rueping, K.-W. Huang, H. Zhang
Adv. Mater. 2025, 37, 2411813
DOI: 10.1002/adma.202406748

Abstract:
Photosensitizer-assisted photocatalytic systems offer a solution to overcome the limitations of inherent light harvesting capabilities in catalysts. However, achieving efficient charge transfer between the dissociative photosensitizer and catalyst poses a significant challenge. Incorporating photosensitive components into reactive centers to establish well-defined charge transfer channels is expected to effectively address this issue. Herein, the electrostatic-driven self-assembly method is utilized to integrate photosensitizers into metal–organic frameworks, constructing atomically Ru-Cu bi-functional units to promote efficient local electron migration. Within this newly constructed system, the [Ru(bpy)2]2+ component and Cu site serve as photosensitive and catalytic active centers for photocarrier generation and H2O2 production, respectively, and their integration significantly reduces the barriers to charge transfer. Ultrafast spectroscopy and in situ characterization unveil accelerated directional charge transfer over Ru-Cu units, presenting orders of magnitude improvement over dissociative photosensitizer systems. As a result, a 37.2-fold enhancement of the H2O2 generation rate (570.9 µmol g−1 h−1) over that of dissociative photosensitizer system (15.3 µmol g−1 h−1) is achieved. This work presents a promising strategy for integrating atomic-scale photosensitive and catalytic active centers to achieve ultrafast photocarrier transfer and enhanced photocatalytic performance.
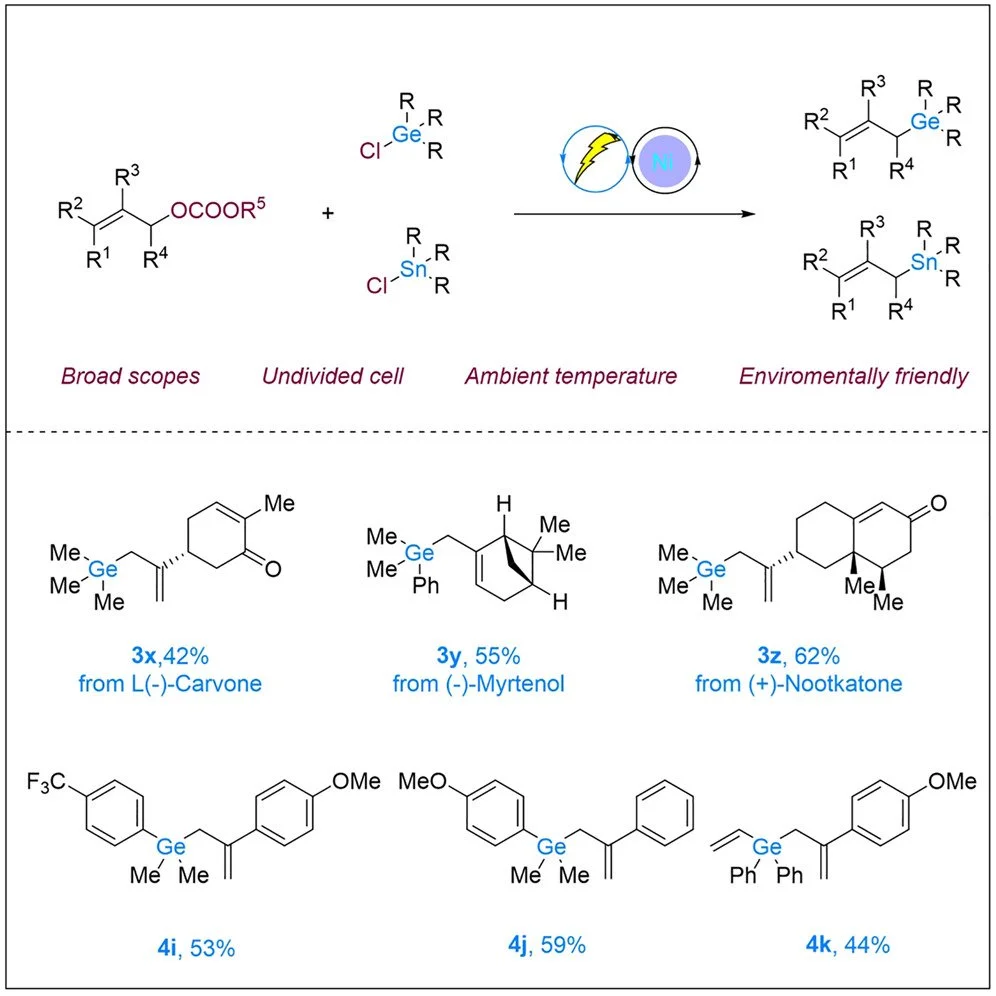
Allylgermane synthesis via facile and general nickela-electrocatalyzed electrophile coupling
Haifeng Chen, Cai Zhai, Chen Zhu, Magnus Rueping
Chem Catalysis, 2025, 101257
DOI: 10.1016/j.checat.2024.101257
Abstract:
Organogermanes have played a significant role in organic chemistry, and effective strategies for accessing various organogermanes are crucial for advancing their applications. However, the formation of allylgermanes under electrophile coupling is still unexplored. Herein, we describe a germylative allylation applying readily accessible allylic carbonates and chlorogermanes. The newly developed method demonstrates good selectivity and high functional group compatibility under mild conditions and provides a variety of allylgermanes, as well as allyl tin, in good yields. Mechanistic and density functional theory (DFT) studies revealed the synergistic catalytic process in detail.
Fine-Tuning NIR-Absorbing BODIPYs for Photoacoustic Detectionof Hypochlorous Ion (OCl⁻)
Prosenjit Isar, Tarushyam Mukherjee, Tengfei Ji, Alexander Nellessen, Jean-Michel Merkes, Fabian Kiessling, Magnus Rueping, Srinivas Banala
Chem. Asian J. 2025, e202401869
DOI: 10.1002/asia.202401869
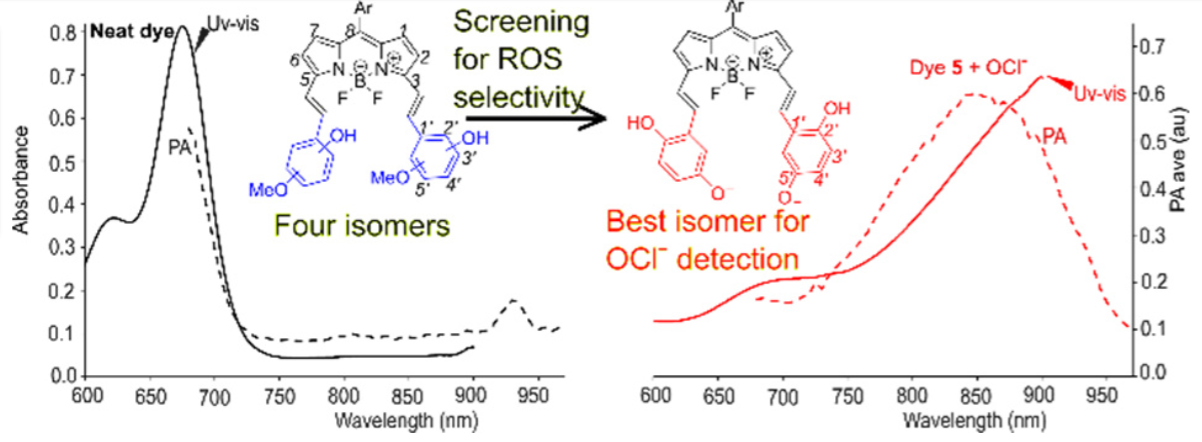
Abstract:
Highly reactive oxygen and nitrogen species (ROS/RNS) play crucial roles in various pathological conditions. Among them, hypochlorous ion (OCl⁻), a potent ROS, is associated in numerous oxidative stress-related disorders. Elevated levels of OCl⁻ are thus recognized as a biomarker for diagnosing inflammatory conditions. To enable selective detection of OCl⁻ via photoacoustic (PA) imaging, we present development of a near infrared(NIR)-absorbing BODIPY-based acoustogenic probe. Four regioisomers of methoxyphenols-conjugated BODIPYs were synthesized to investigate the positional influence on OCl− selectivity over other ROS/RNS. Our study reveals that only one isomer, 4-methoxy phenol conjugation, exhibited exceptional selectivity for OCl− without any competitive reactions, making it suitable for PA imaging. This study highlights the importance of regioisomers characterization in achieving intricate selectivity among competing reactive species. The fine-tuning and development of a suitable dye now enable the optimization of physicochemical properties for in vivo OCl− detection using PA imaging.
Tracking Water Splitting Activity by Cocatalyst Identity in SrTiO3
Nursaya Zhumabay, Jeremy A. Bau, Rafia Ahmad, Laurentiu Braic, Huabin Zhang, Luigi Cavallo, Magnus Rueping
Small Struct. 2025, 6, 2400283
DOI: 10.1002/sstr.202400283

Abstract:
Photocatalytic water splitting is the most idealistic route to green hydrogen production, but the extensive material requirements for this reaction make it difficult to realize good photocatalysts. Noble metal cocatalysts are often added to photocatalysts to aid in charge separation and improve surface kinetics for H2 evolution. In this study, the high activity of the promising photocatalyst Al-doped SrTiO3 is demonstrated to be ultimately dependent on the cocatalyst used as much as the presence of Al dopant. By tracking the band energetics of photocatalyst electrodes using operando electrochemical ATR-SEIRAS, cocatalysts (especially Rh) are found to shift the quasi-Fermi levels and metal-semiconductor flat-band potentials of photocatalysts in an anodic direction. Furthermore, the size of the shift directly correlates with overall water splitting activity, demonstrating that SrTiO3 becomes more active as photo-generated electrons are stabilized further from the conduction band. Rh on Al-doped SrTiO3 provides the most advantageous band tailoring as confirmed by DFT, and is experimentally found to provide this effect by eliminating Ti3+-related surface traps in the presence of Al dopants. Therefore, the effect of cocatalysts on water splitting activity is more complicated than previously thought.
Anton S. Makarov, Magnus Rueping
Green Chem. 2025, 26, 716-721
DOI: 10.1039/d4gc04623d

Abstract:
This study presents a scalable mechanochemical method for the upcycling of (poly)lactic acid (PLA) into industrially valuable alkyl lactate esters via organocatalytic depolymerizing transesterification enabled by resonant acoustic mixing (RAM). The process is characterized by its simplicity, requiring neither grinding media nor a co-solvent and utilizing nearly stoichiometric amounts of an alcohol reaction partner in the presence of an inexpensive, easily accessible catalyst. Additionally, the mechanochemistry is successfully extended to the upcycling of post-consumer PLA for the synthesis of various substituted esters and lactamides.
Ba/Ti MOF: A Versatile Heterogeneous Photoredox Catalyst for Visible-Light Metallaphotocatalysis
I. S. Khan, L. Garzon-Tovar, R. Kancherla, N. Kolobov, A. Dikhtiarenko, M. Almalki, A. Shkurenko, V. Guillerm, K. N. Le, G. Shterk, C. H. Hendon, M. Eddaoudi, J. Gascon, M. Rueping
Adv. Mater. 2025, 37, 2405646
DOI: 10.1002/adma.202405646

Abstract:
The field of sustainable catalysis is evolving rapidly, with a strong emphasis on developing catalysts that enhance efficiency. Heterogeneous photocatalysis has emerged as a promising and mild approach to address the persistent challenges. Among various heterogeneous photocatalysts, Metal-Organic Frameworks (MOFs) have gained significant attention for their exceptional performance and recyclability in photocatalytic reactions. In this context, contrary to the conventional homogeneous Ir or Ru-based photocatalysts, which face significant challenges in terms of availability, cost, scalability, and recyclability, we have developed a new Ba/Ti MOF (ACM-4) as a heterogeneous catalyst that could mimic/outperform the conventional photocatalysts, offering a more sustainable solution for efficient chemical processes. Its redox potential and triplet energy are comparable to or higher than the conventional catalysts, organic dyes, and metal semiconductors, enabling its use in both electron transfer and energy transfer applications. It facilitates a broad range of coupling reactions involving pharmaceuticals, agrochemicals, and natural products, and is compatible with various transition metals such as Ni, Cu, Co, and Pd as co-catalysts. Notably, ACM-4 can be easily recovered and reused multiple times with minimal loss in efficiency. The effectiveness of the ACM-4 as a photocatalyst is supported by comprehensive material studies and photophysical experiments. These significant findings underscore the potential of ACM-4 as a highly versatile and cost-effective photoredox catalyst, providing a sustainable, one-material solution for efficient chemical processes.