
2018
24. Sustainable Alkylation of Unactivated Esters and Amides with Alcohols Enabled by Manganese Catalysis
Yoon Kyung Jang, Tobias Krückel, Magnus Rueping, Osama El-Sepelgy
Org. Lett. 2018, 20, 7779-7783

Abstract:
The first example of manganese-catalyzed C-alkylation of the carboxylic acid derivatives is reported. The bench-stable homogeneous manganese complex enables the transformation of the renewable alcohol and carboxylic acid derivative feedstock to higher value esters and amides. The reaction operates via hydrogen autotransfer and ideally produces water as the only side product. Importantly, aliphatic-, benzylic-, and heterocyclic-containing alcohols can be used as alkylating reagents, eliminating the need for mutagenic alkyl halides.
Krishnamoorthy Muralirajan, Rajesh Kancherla, Magnus Rueping
Angew. Chem. Int. Ed. 2018, 57, 14787-14791

Abstract:
An unprecedented dehydrogenative aromatization sulfonylation reaction of pyrrolidines was achieved under photoredox catalysis with the use of sulfonyl chlorides which have a dual function as catalyst regeneration and sulfonylation reagent. The isolation of the intermediates provided insight into the reaction mechanism of this new aromatization protocol. N-Alkyl pyrrolides and in particular N-benzyl-protected derivatives can be effectively applied and that the possible formation of exocyclic imines resulting in aldehydes or ketones cannot be observed. The full dehydrogenation of saturated heterocycles is a topical area in synthesis as well as in hydrogen storage and release but typically requires the use of transition metals and/or higher temperatures. The new protocol, however, allows the dehydrogenative aromatization of pyrrolidine without the need for additional transition metals or oxidants; proceeds at room temperature; and thus exhibits excellent functional group tolerance. Moreover, the regioselective sulfonylation provides β-substituted sulfonyl pyrroles which are otherwise difficult to access and which can be used in the synthesis of new nitrogen/sulfon-fused, ladder-type, π-conjugated molecules.
Viktoriia Zubar, Yury Lebedev, Luis Miguel Azofra, Luigi Cavallo, Osama El‐Sepelgy, Magnus Rueping
Angew. Chem. Int. Ed. 2018, 57, 13439-13443

Abstract:
The first base-metal-catalysed hydrogenation of CO2-derived carbonates to alcohols is presented. The formal CO2 to methanol reaction proceeds under mild conditions in the presence of a well-defined manganese complex with a loading as low as 0.25 mol %. The non-precious-metal homogenous catalytic system provides an indirect route for the conversion of CO2 into methanol with the co-production of value-added (vicinal) diols in yields of up to 99 %. Experimental and computational studies indicate a metal–ligand cooperative catalysis mechanism.
Yunfei Cai, Yurong Tang, Lulu Fan, Quentin Lefebvre, Hong Hou, Magnus Rueping
ACS Catal. 2018, 8, 9471-9476

Abstract:
A first protocol for the photooxidative activation of α-silylamines and α-amino acids for desilylative and decarboxylative additions, allylations, and heteroarylations in the presence of mesoporous graphitic carbon nitride (mpg-CN) was developed. The procedure has broad scope and provides the desired products in high yields. The heterogeneous nature of the g-C3N4 catalytic system enables easy recovery and recycling as well as the use in multiple runs without loss of activity. The photoredox catalyzed reactions can also be conducted in continuous photo flow fashion and scaled up to gram-scale. Thus, the stable and readily available polymeric g-C3N4 provides an alternative to homogeneous photosensitizers for the generation of valuable radical intermediates for applications in synthesis and catalysis.
20. Nickel-Catalyzed Csp2–Csp3 Bond Formation via C–F Bond Activation
Yee Ann Ho, Matthias Leiendecker, Xiangqian Liu, Chengming Wang, Nurtalya Alandini, Magnus Rueping
Org. Lett. 2018, 20, 5644-5647

Abstract:
A direct method for the Csp2–Csp3 bond formation with aryl fluorides as electrophiles has been developed. By using a Ni(COD)2/dppe catalytic system, the alkylation of aryl fluorides could be performed with various alkyl Grignard reagents with yields of up to 99%. The established methodology proved successful in avoiding the undesired β-hydride elimination and is appealing in terms of electrophile availability as no directing or activating groups are required. Moreover, the components of the catalytic system (Ni(COD)2 and dppe) and the Grignard reagents are readily available. In addition, the use of LiCH2SiMe3 as nucleophile resulted in products that can be converted into more complex structures by taking advantage of the existing SiMe3 group.
Jiaqi Jia, Yee Ann Ho, Raoul F. Bülow, Magnus Rueping
Chem. Eur. J. 2018, 24, 14054-14058

Abstract:
A new method for the synthesis of alkyne- and alkene-decorated lactams by employing a Brønsted base assisted, visible-light, photoredox-catalyzed, intramolecular 5-exo-trig cyclization/intermolecular radical addition/elimination reaction was developed. The feasibility to retain functional groups on the radical acceptor by introducing a sulfonyl group is successfully proven by the products obtained. The method developed is widely applicable, affording products in good to high yields for a broad range of substrates, with high functional group tolerance starting from readily available amides. This provides the first reported access to these lactam and oxazolidinone core structures, which will be useful for further functionalization into valuable fine chemicals.
18. Room-Temperature C–H Bond Functionalization by Merging Cobalt and Photoredox Catalysis
Deepti Kalsi, Subhradeep Dutta, Nagaraju Barsu, Magnus Rueping, Basker Sundararaju
ACS Catal. 2018, 8, 8115-8120

Abstract:
A non-noble metal-free protocol has been developed for C–H bond functionalization at room temperature by merging cobalt-mediated catalysis with photocatalysis. The reaction requires only oxygen as sole oxidant and operated at room temperature under redox-neutral conditions. Visible-light activated photoredox catalyst functions as an electron transfer reagent with oxygen as a terminal oxidant in the cobalt-mediated C–H and N–H bond annulation. The developed methodology allows annulations with various coupling partners. The concept demonstrated herein is expected to enhance the scope of cobalt catalysis as applied to sustainable fine chemical synthesis.
Ruediger Borrmann, Volodymyr Palchyk, Andrij Pich, Magnus Rueping
ACS Catal. 2018, 8, 7991-7996

Abstract:
A switchable colloidal catalysts based on temperature-responsive polymer microgels that combine the advantages of homogeneous and heterogeneous catalysts has been developed. The new adaptable catalyst system consists of a porous microgel structure in which a covalently attached organocatalyst acts as a catalysis center mimicking the active site of an enzyme. In comparison to enzymes, the temperature responsiveness of the microgel leads to distinct advantages. The catalyst activity can be simply reversibly switched on and off by adjustment of the temperature. At reaction temperature (T > VPTT) the microgels are swollen and colloidally stable, which ensures a large surface area, high reaction speed, and selectivity. With a decrease in temperature to T < VPTT the microgels collapse and form aggregates in solution and the catalytic activity is switched off. The colloidal aggregates can be easily separated from the reaction mixture by filtration or centrifugation; hence, no further workup is needed. The catalyst can be recycled several times as the microgel aggregates solubilize again to provide the active catalyst system. This provides a unique possibility of fast recovery and reuse of the colloidal microgel catalyst, which results in the lowering of the production costs and reduction of the product pollution risk often observed in metal-catalyzed processes. In addition, microgel-based catalysts exhibit superior thermal and chemical resistance. As a representative example we demonstrated the application of this temperature-responsive enzyme-mimicking microgel containing a bifunctional quinidylsulfomamide moiety as the active catalysis site in the desymmetrization of meso-anhydrides. Under reaction conditions, above its UCST, the catalytically active microgel is swollen and well solvated and therefore acts as a homogeneous catalyst with advantages of high reaction yield (>95%) and good enantiocontrol. After the reaction has finished, the catalyst can be switched off by simply decreasing the temperature below the UCST, which leads to shrinking, aggregation, and precipitation of the microgel. We performed several subsequent catalytic cycles by cooling the reaction mixture and separating the precipitated microgel by filtration. The experimental data indicate that the high efficiency and selectivity of the colloidal catalyst remains unaltered after many separation cycles. Thus, the interesting properties of these new switchable and recyclable microgel-based catalysts open up new possibilities and opportunities for designing and preparing new adaptable organic nanoreactors with high efficiency.
Long Huang, Magnus Rueping
Angew. Chem. Int. Ed. 2018, 57, 10333-10337

Abstract:
A direct regioselective coupling of allylic C(sp3)−H bonds with aryl- and vinylbromides enabled by the combination of nickel and photoredox catalysis, provides an unprecedented method for the construction of allylarenes, as well as 1,4-dienes from unactivated alkenes under very mild reaction conditions. The exclusive preference for primary allylic C(sp3)−H bonds can be rationalized by a hydrogen atom transfer (HAT) abstraction process with photocatalytically generated bromine radical. Investigations regarding the further application of this new method as well as experiments to gain a more detailed understanding of the reaction mechanism are currently part of our research.
15. Rhenium‐ and Manganese‐Catalyzed Selective Alkenylation of Indoles
Chengming Wang, Magnus Rueping
ChemCatChem 2018, 10, 2681-2685

Abstract:
An efficient rhenium-catalyzed regioselective C−H bond alkenylation of indoles is reported. The protocol operates well for internal as well as terminal alkynes, affording products in good to excellent yields. Furthermore, a manganese-catalyzed, acid free, regioselective C2-alkenylation of indoles with internal alkynes is described. The directing groups can be easily removed after the reaction and the resulting products can be used as valuable building blocks for the synthesis of diverse heterocyclic compounds.
Lin Guo, Magnus Rueping
Chem. Eur. J. 2018, 24, 7794-7809

Abstract:
Owing to the unique advantages of carbonyl functionalities, chemists have recently developed a series of novel exciting protocols that have not been achieved with traditional methods before. In this Review, we have tried to emphasize the recent progress in the field of transition-metal-catalyzed decarbonylative coupling reactions in which a series of carbonyl functional group substitutions have been successfully achieved, including decarbonylative arylation, alkylation, olefination, alkynylation, cyanation, etherification, amination, silylation, borylation, thiolation, phosphination, stannylation, reduction and retro-hydrocarbonylation. This synthetic approach is desired to offer new opportunities for organic synthesis. But, still, some interesting yet challenging problems remain unsolved. The use of unactivated aliphatic aroyl derivatives in decarbonylative cross-coupling is still a challenge. Often high temperatures are used which restrict industrial applications. Nevertheless, the developments stand for a great advance in the area of functional group interconversion and expand the repertoire of synthetic methods which can be considered in late stage functionalizations as well as synthesis and retrosynthesis in general.
13. Multiple Hydrogen‐Bond Activation in Asymmetric Brønsted Acid Catalysis
Hsuan‐Hung Liao, Chien‐Chi Hsiao, Iuliana Atodiresei, Magnus Rueping
Chem. Eur. J. 2018, 24, 7718-7723

Abstract:
Herein we describe an efficient enantioselective cycloaddition reaction for the synthesis of various multisubstituted optically enriched tetrahydroquinolines (THQs) by means of asymmetric Brønsted acid catalysis. A chiral 1,1′-spirobiindane-7,7′-diol (SPINOL)-derived N-triflylphosphoramide (NTPA) was able to generate aza-ortho-quinone methides (aza-o-QMs) in situ and to promote their subsequent asymmetric cycloaddition with unactivated alkenes to afford valuable THQ derivatives bearing two or three stereogenic centers in excellent yields with excellent enantioselectivities. Particularly noteworthy are the high tolerance toward a broad range of aza-o-QMs and alkenes and the fact that all cycloadducts were obtained as single diastereomers. Furthermore, valuable optically enriched Julolidine analogues could also be obtained by using this protocol.26 Given the wide applicability of aza-o-QMs in organic synthesis and the many possibilities for the extension of this mild asymmetric catalysis protocol to other cycloadditions as well as nucleophilic additions, we believe that this work is a good basis to stimulate further work and applications of this developed catalysis methodology.
12. Cross-Coupling of Amides with Alkylboranes via Nickel-Catalyzed C–N Bond Cleavage
Xiangqian Liu, Chien-Chi Hsiao, Lin Guo, Magnus Rueping
Org. Lett. 2018, 20, 2976-2979

Abstract:
We have developed a versatile nickel-catalyzed alkylation of amides with alkylboranes as nucleophilic counterparts. The N-Ph, Me amides were cleaved under mild conditions and coupled with a range of functionalized alkylboranes with high efficiency. The catalytic alkylation proceeded selectively at the C–N bond of N-Ph, Me amides in the presence of ester and N-alkyl, alkyl amide groups. The good chemoselectivity exhibited by our deamidative alkylation protocol may not be achieved by applying the traditional ketone synthesis from amides in which typically strong organometallic reagents have to be used.
11. Manganese Catalyzed Regioselective C–H Alkylation: Experiment and Computation
Chengming Wang, Bholanath Maity, Luigi Cavallo, Magnus Rueping
Org. Lett. 2018, 20, 3105-3108
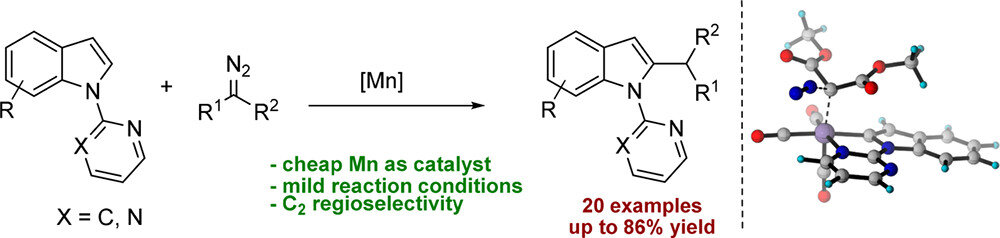
Abstract:
A new efficient manganese-catalyzed selective C2-alkylation of indoles via carbenoid insertion has been achieved. The newly developed manganese catalyzed C–H functionalization protocol provides access to diverse products and shows good functional group tolerance. Mechanistic and computational studies support the formation of a Mn(CO)3 acetate complex as the catalytically active species.
Lin Guo, Magnus Rueping
Acc. Chem. Res. 2018, 51, 1185-1195
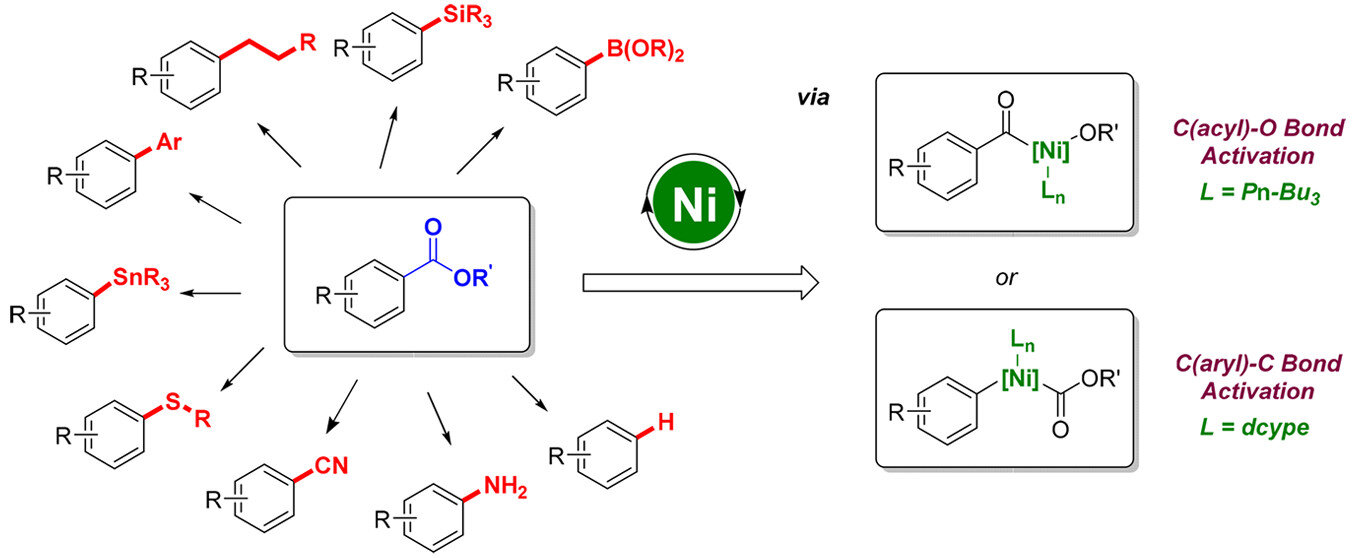
The utilization of carboxylic acid esters as electrophiles in metal-catalyzed cross-coupling reactions is increasingly popular, as environmentally friendly and readily available ester derivatives can be powerful alternatives to the commonly used organohalides. However, key challenges associated with the use of these chemicals remain to be addressed, including the stability of ester substrates and the high energy barrier associated with their oxidative addition to low-valent metal species. Due to recent developments in nickel catalysis that make it easier to perform oxidative additions, chemists have become interested in applying less reactive electrophiles as coupling counterparts in nickel-catalyzed transformations. Hence, our group and others have independently investigated various ester group substitutions and functionalizations enabled by nickel catalysis. Such methods are of great interest as they enable the exchange of ester groups, which can be used as directing groups in metal-catalyzed C-H functionalizations prior to their replacement. Here, we summarize our recent efforts toward the development of nickel-catalyzed decarbonylative cross-coupling reactions of carboxylic esters. Achievements accomplished by other groups in this area are also included. To this day, a number of new transformations have been successfully developed, including decarbonylative arylations, alkylations, cyanations, silylations, borylations, aminations, thioetherifications, stannylations, and hydrogenolysis reactions. These transformations proceed via a nickel-catalyzed decarbonylative pathway and have shown a high degree of reactivity and chemoselectivity, as well as several other unique advantages in terms of substrate availability, due to the use of esters as coupling partners. Our combined experimental and computational study of a ligand-controlled chemoselective nickel-catalyzed cross-coupling of aromatic esters with alkylboron reagents provided further insight into the reaction mechanism. We demonstrated that nickel complexes with bidentate ligands favor the C(aryl)-C bond cleavage in the oxidative addition step, resulting in decarbonylative alkylations, while nickel complexes with monodentate phosphorus ligands promote the activation of the C(acyl)-O bond, leading to the production of ketone products. Although more detailed mechanistic investigations need to be undertaken, the successful development of decarbonylative cross-coupling reactions can serve as a solid foundation for future studies. We believe that this type of decarbonylative cross-coupling reactions will be of significant value, in particularly in combination with the retrosynthetic analysis and synthesis of natural products and biologically active molecules. Thus, the presented ester substitution methods will pave the way for successful applications in the construction of complex frameworks by late-stage modification and functionalization of carboxylic acid derivatives.
Aleksandra Brzozowska, Luis Miguel Azofra, Viktoriia Zubar, Iuliana Atodiresei, Luigi Cavallo, Magnus Rueping, Osama El-Sepelgy
ACS Catal. 2018, 8, 4103-4109
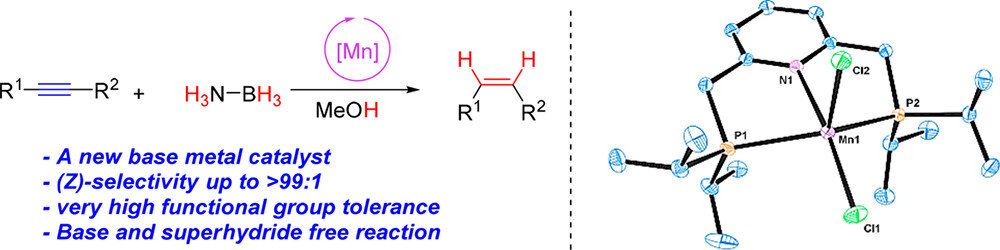
Abstract:
Herein we report unprecedented manganese-catalyzed semihydrogenation of internal alkynes to (Z)-alkenes using ammonia borane as a hydrogen donor. The reaction is catalyzed by a pincer complex of the earth-abundant manganese(II) salt in the absence of any additives, base, or superhydride. The ammonia borane smoothly reduces the manganese precatalyst [Mn(II)–PNP][Cl]2 to the catalytically active species [Mn(I)–PNP]–hydride in the triplet spin state. This manganese hydride is highly stabilized by complexation with the alkyne substrate. Computational density functional theory (DFT) analysis studies of the reaction mechanism rationalize the origin of stereoselectivity toward formation of (Z)-alkenes.
Hong Hou, Shaoqun Zhu, Iuliana Atodiresei, Magnus Rueping
Eur. J. Org. Chem. 2018, 2018, 1277-1280

Abstract:
We have developed a combined catalytic system for the highly enantio- and diastereoselective α-alkylation of tetrahydroisoquinolines. In the present dual catalytic protocol ketones are activated by a chiral primary amine catalyst and tetrahydroisoquinolines are activated by a visible-light photoredox catalyst. The desired α-alkylation products were obtained in good yields, with high enantio- and diastereoselectivity. Studies on further challenging asymmetric reactions combining organo- and photoredox catalysis are currently underway in our laboratories.
Adisak Chatupheeraphat, Hsuan-Hung Liao, Watchara Srimontree, Lin Guo, Yury Minenkov, Albert Poater, Luigi Cavallo, Magnus Rueping
J. Am. Chem. Soc. 2018, 140,3724-3735
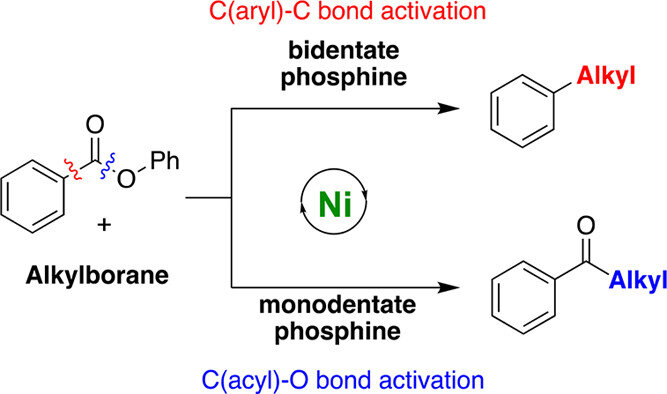
Abstract:
A ligand-controlled and site-selective nickel catalyzed Suzuki–Miyaura cross-coupling reaction with aromatic esters and alkyl organoboron reagents as coupling partners was developed. This methodology provides a facile route for C(sp2)–C(sp3) bond formation in a straightforward fashion by successful suppression of the undesired β-hydride elimination process. By simply switching the phosphorus ligand, the ester substrates are converted into the alkylated arenes and ketone products, respectively. The utility of this newly developed protocol was demonstrated by its wide substrate scope, broad functional group tolerance and application in the synthesis of key intermediates for the synthesis of bioactive compounds. DFT studies on the oxidative addition step helped rationalizing this intriguing reaction chemoselectivity: whereas nickel complexes with bidentate ligands favor the C(aryl)–C bond cleavage in the oxidative addition step leading to the alkylated product via a decarbonylative process, nickel complexes with monodentate phosphorus ligands favor activation of the C(acyl)–O bond, which later generates the ketone product.
Shao‐Chi Lee, Hsuan‐Hung Liao, Adisak Chatupheeraphat, Magnus Rueping
Chem. Eur. J. 2018, 24, 3608-3612

Abstract:
We have developed a new and selective Nickel catalyzed cross-coupling reaction of esters and amides with mercaptans as coupling partners. The transformation of aryl, alkenyl and alkyl esters/amides to the corresponding thioethers was previously not known and is, with conventional methods, difficult to achieve. Thus, the newly developed methodology enables a facile route for C−S bond formation in a straightforward fashion. Various mercaptans and a wide range of ester and amide substrates bearing various substituents are tolerated in this process which afforded products in good to excellent yields. In addition, an intramolecular version of this reaction has also been developed. The utility of this newly designed protocol has been demonstrated in the synthesis of benzothiophene on the bench top. Given the simplicity and generality of the protocol it is anticipated that it should find application in synthesis, retrosynthesis and late stage modification.
Roman Pluta, Patricia E. Krach, Luigi Cavallo, Laura Falivene, Magnus Rueping
ACS Catal. 2018, 8, 2582-2588

Abstract:
A new iodoarene catalyst for metal-free asymmetric fluorination of carbonyl compounds was developed. Notably, the presented methodology utilizes inexpensive reagents including hydrogen fluoride as fluorine source to generate the catalytically active iodoarene difluoride species in the presence of mCPBA as oxidant. The reactions proceed at room temperature under air, tolerate a wide range of functional groups, and give products with excellent enantioselectivities. Mechanistic experiments imply the importance of a hydrogen fluoride-containing complex for catalytic activity, which is important for the design of further catalytic asymmetric fluorinations as well as iodoarene-catalyzed reactions. DFT calculations provided insight on the whole reaction mechanism and on the origin of enantioselectivity.
Osama El-Sepelgy, Aleksandra Brzozowska, Jan Sklyaruk, Yoon Kyung Jang, Viktoriia Zubar, Magnus Rueping
Org. Lett. 2018, 20, 696-699

Abstract:
Herein, we report the first example of an iron catalyzed intramolecular hydroamination of allenic amines to enamines as well as an efficient base metal catalyzed hydroalkoxylation of α-allenic alcohols to valuable cyclic enol ethers. The general applicability of this protocol is highlighted by the synthesis of 30 unsaturated heterocycles with good yields and excellent chemoselectivity. The key to achieve this unusual selectivity is the use of an iron-based metal–ligand catalyst which can activate the substrate in a dual catalytic fashion. The absence of any sensitive chemicals as well as base additives is an additional advantage of the reported protocol. We believe that this strategy will find practical application in the synthesis of bioactive molecules and natural products.
Huifeng Yue, Chen Zhu, Magnus Rueping
Angew. Chem. Int. Ed. 2018, 57, 1371-1375

Abstract:
We describe a novel and efficient method for the synthesis of sulfones by photoredox/nickel catalysis for the first time. This protocol allows the cross-coupling of a series of sodium sulfinates with a wide range of aryl, heteroaryl, and vinyl bromides and iodides, as well as more challenging aryl chlorides. Importantly neither sacrificial reagents nor organic electron mediators are necessary in this reaction. Moreover, the utility of sodium sulfinates as precursors of sulfonyl radicals and the generation of reactive NiIII intermediates promote this transformation at room temperature. Therefore, the reaction possesses a broad tolerance of functional groups, showing its advantages in comparison to the traditional methods.
Huifeng Yue, Chen Zhu, Magnus Rueping
Org. Lett. 2018, 20, 385-388

Abstract:
The functional group interconversion of inert methyl esters into stannanes was realized for the first time with the aid of nickel catalysis. A series of methyl esters as well as other common esters, such as ethyl, cyclohexyl, benzyl and phenyl esters, all underwent this decarbonylative stannylation protocol smoothly, affording diverse arylstannanes which are important in the construction of C–C and C–heteroatom bonds. Moreover, this method could efficiently differentiate between two different types of esters. In addition, the monostannylation/fluorination and monostannylation/arylation of bis-methyl esters could be realized, showing its great practical value. Furthermore, the stannylation protocol shows good chemoselectivity and functional groups including groups previously used in cross-couplings remain intact. Given that arylstannanes are highly valuable products for different applications, this new ester to stannane interconversion will be of value and may become a good alternative to aryl halide stannylation reactions. Efforts to investigate the mechanism and to broaden the scope further are currently ongoing in our laboratories and will be reported in due course.
Stefan W. Grötzinger, Ram Karan, Eva Strillinger, Stefan Bader, Annika Frank, Israa S. Al Rowaihi, Anastassja Akal, Wiebke Wackerow, John A. Archer, Magnus Rueping, Dirk Weuster-Botz, Michael Groll, Jörg Eppinger, Stefan T. Arold
ACS Chem. Biol. 2018, 13, 161-170

Abstract:
Single Amplified Genomes (SAGs) generate a trove of undescribed proteins, including enzymes from unculturable extremophiles. These molecules may combine exceptional stability with unusual functions and could therefore inspire novel biotechnological applications. Nonetheless, this source of proteins has not yet been used for targeted experimental investigations on the protein level, because of a lack of trust in the sequence data, difficulties of the data mining processes, and difficulties with protein expression. (26) The methodology reported here enabled the functional and structural analysis of a SAG-derived archaeal halo-thermophilic ADH. Our experimental characterization of ADH/D1 revealed a unique environmental adaptation to use Mn2+ as a cofactor and a rare cofactor dihydroxy modification under aerobic conditions. The structural analysis suggested a mechanism for this modification, enabled by a nearby histidine. The structural comparison to homologues allowed the features that confer the exceptional stability of ADH/D1 to be identified. The roadmap and proof-of-concept we provide here may alter the way enzymes are obtained, away from the traditional culture- and activity-based methods toward new and more reliable annotation methods. In this view, SAGs, but not metagenomics, offer the possibility of studying many enzymes from the same species. Hence, SAGs could allow for the reconstruction of multiprotein complexes or metabolic pathways and provide genes for one-pot multistep reactions involving several enzymes.