
2016
Lin Guo, Xiangqian Liu, Christoph Baumann, Magnus Rueping
Angew. Chem. Int. Ed. 2016, 55, 15415-15419

Abstract:
We describe the first alkylation reaction of polycyclic aromatic methyl ethers, which is based on the use of alkylboron reagents as coupling partners and involves the cleavage of highly inert C−OMe bonds. This reaction shows broad functional group tolerance, providing a practical and versatile way to form various C(sp2)−C(sp3) bonds that does not suffer from β-hydride elimination. This enables the synthesis of various products in high yields. Diverse synthetically readily accessible and naturally occurring methoxy arenes were efficiently transformed into a variety of alkyl-substituted molecules. As such, this process will be of interest for the replacement of methoxy groups, for example, after their use as directing groups in Friedel–Crafts reactions, ortho lithiations, or metal-catalyzed C−H functionalizations. In retrosynthesis planning, methoxy groups can thus be considered as directing groups and, at the same time, as placeholders for alkyl groups. Furthermore, this method is applicable to a range of methyl enol ethers, providing access to valuable alkyl vinylarenes.
Yunfei Cai, Yurong Tang, Iuliana Atodiresei, Magnus Rueping
Angew. Chem. Int. Ed. 2016, 55, 14126-14130

Abstract:
We have developed the first catalytic asymmetric Piancatelli reaction. The Brønsted acid catalyzed reaction shows broad substrate scope and proceeds well with various amine nucleophiles, as well as secondary and tertiary carbinols, to give a range of 4-aminocyclopentenones (60 examples) in good yields and with excellent diastereo- and enantiocontrol. Under the mild reaction conditions, even alkyl-substituted tertiary furylcarbinols, which previously proved to be challenging substrates for Piancatelli reactions, were tolerated and provided the desired products with a quaternary stereocenter. To demonstrate the value of our new approach, the protocol was applied to the enantioselective synthesis of a cyclopentane-based hNK1 antagonist. The aza-Piancatelli reaction reported here not only provides fast access to valuable compounds but also constitutes a good basis for the development of further Brønsted acid catalyzed rearrangements and electrocyclization reactions
Lulu Fan, Jiaqi Jia, Hong Hou, Quentin Lefebvre, Magnus Rueping
Chem. Eur. J. 2016, 22, 16437-16440

Abstract:
A mild decarboxylative aminomethylation of aryl- and vinyl sulfonates using a combination of visible-light photoredox and nickel catalysis through a C−O bond-cleavage procedure is described for the first time. The reaction has broad substrate scope and aryl- triflates, mesylates, tosylates, and alkenyl triflates can be applied to give the desired aminoalkylated products and benzyl-protected anilines in good to excellent yields. The reaction conditions, allowing the cross-coupling to occur under visible light at room temperature in the presence of various functional groups, are rather mild and can be selectively performed or even be extended to chlorinated arenes if needed. Furthermore, we demonstrate that phosphine complexes can be used in the dual catalytic procedure calling for the development of combined metal and photoredox-catalyzed asymmetric reactions.
Lin Guo, Magnus Rueping
Chem. Eur. J. 2016, 22, 16787-16790

Abstract:
The first nickel-catalyzed decarbonylative borylation reaction of carboxylic acid esters is described herein. This protocol provides a new possibility to cleave a stable ester group while forming a versatile boron group. The broad substrate scope, high reactivity, and scalability of the method suggest that this protocol can be a powerful alternative to the existing methodologies for preparing organoboron compounds from readily available precursors. It is important to note that the newly developed protocol shows high chemoselectivity and that functional groups such as C−OMe, C−F, and C−NCOR, previously used in nickel-catalyzed functional-group interconversions remain intact. Thus, the developed protocol will be of interest as it allows for the replacement of ester groups, which can be used prior to the replacement as a directing group in Friedel–Crafts reactions or metal-catalyzed C−H functionalizations. The method may also be considered in retrosynthesis planning, using the ester group as a directing group and, at the same time, as placeholder for the boron functionality, which, if required, can be easily functionalized to other valuable products.
Osama El‐Sepelgy, Nurtalya Alandini, Magnus Rueping
Angew. Chem. Int. Ed. 2016, 55, 13602-13605

Abstract:
We report the first example of a cooperative nonnatural iron/lipase catalytic system. The key to success is the unprecedented combination and compatibility of a reactive iron dehydrogenation–hydrogenation catalyst with a lipase. The iron-catalyzed hydrogen autotransfer proceeds under very mild reaction conditions which facilitates the combination with the enzyme. Various racemic alcohols including benzylic, aliphatic, and heteroaromatic alcohols have been transformed into enantioenriched acetates without the use of precious metal catalysts and expensive chiral ligands. Given the compatibility of iron catalysis with enzymatic resolutions demonstrated here and the fact that the iron catalyst is inexpensive and readily available, we believe that these initial promising findings will open up new avenues for further exploration of this effective dual catalysis system.
Lin Guo, Adisak Chatupheeraphat, Magnus Rueping
Angew. Chem. Int. Ed. 2016, 55, 11810-11813

Abstract:
Copper and nickel: An efficient nickel/copper-catalyzed decarbonylative silylation reaction of carboxylic acid esters with silylboranes is described. This reaction provides access to structurally diverse silanes with high efficiency and excellent functional-group tolerance starting from readily available esters. An efficient nickel/copper-catalyzed decarbonylative silylation reaction of carboxylic acid esters with silylboranes is described. This reaction provides access to structurally diverse silanes with high efficiency and excellent functional-group tolerance starting from readily available esters.
Anthony Millet , Quentin Lefebvre, Magnus Rueping
Chem. Eur. J. 2016, 22, 13464-13468

Abstract:
We have successfully developed a tin-free and halide-free Giese reaction by efficiently generating primary and secondary α-amino radicals starting from readily accessible amino acids. The α-amino radicals could be efficiently coupled with various α,β-unsaturated carbonyl compounds to give various cyclic and acyclic γ-amino ketones with different substitution patterns. Furthermore, we were able to demonstrate the possibility to apply this method to N-heterocyclic amino acids which allows the access to various biologically active molecules.
David C. Fabry, Magnus Rueping
Acc. Chem. Res. 2016, 49, 1969-1979

Abstract:
The development of efficient catalytic systems for direct aromatic C–H bond functionalization is a long-desired goal of chemists, because these protocols provide environmental friendly and waste-reducing alternatives to classical methodologies for C–C and C–heteroatom bond formation. A key challenge for these transformations is the reoxidation of the in situ generated metal hydride or low-valent metal complexes of the primary catalytic bond forming cycle. To complete the catalytic cycle and to regenerate the C–H activation catalyst, (super)stoichiometric amounts of Cu(II) or Ag(I) salts have often been applied. Recently, “greener” approaches have been developed by applying molecular oxygen in combination with Cu(II) salts, internal oxidants that are cleaved during the reaction, or solvents or additives enabling the metal hydride reoxidation. All these approaches improved the environmental friendliness but have not overcome the obstacles associated with the overall limited functional group and substrate tolerance. Hence, catalytic processes that do not feature the unfavorable aspects described above and provide products in a streamlined as well as economically and ecologically advantageous manner would be desirable. In this context, we decided to examine visible light photoredox catalysis as a new alternative to conventionally applied regeneration/oxidation procedures. This Account summarizes our recent advances in this expanding area and will highlight the new concept of merging distinct redox catalytic processes for C–H functionalizations through the application of visible light photoredox catalysis. Photoredox catalysis can be considered as catalytic electron-donating or -accepting processes, making use of visible-light absorbing homogeneous and heterogeneous metal-based catalysts, as well as organic dye sensitizers or polymers. As a consequence, photoredox catalysis is, in principle, an ideal tool for the recycling of any given metal catalyst via a coupled electron transfer (ET) process. Here we describe our first successful endeavors to address the above challenges by combining visible light photoredox catalysis with different ruthenium, rhodium, or palladium catalyzed C–H activations. Since only small amounts of the oxidant are generated and are immediately consumed in these transformations, side reactions of substrates or products can be avoided. Thus, usually oxidant-sensible substrates can be used, which makes these methods highly suitable for complex molecular structure syntheses. Moreover, mechanistic studies shed light on new reaction pathways, intermediates, and in situ generated species. The successful development of our dual catalysis concept, consisting of combined visible light photoredox catalysis and metal catalyzed C–H functionalization, provides many new opportunities for further explorations in the field of C–H functionalization.
9. Photoredox-Catalyzed Ketyl–Olefin Coupling for the Synthesis of Substituted Chromanols
Eleonora Fava, Masaki Nakajima, Anh L. P. Nguyen, Magnus Rueping
J. Org. Chem. 2016, 81, 6959-6964

Abstract:
We report a ketyl–olefin coupling for the preparation of substituted 3-benzylchroman-4-ols, promoted by visible light photoredox catalysis. Importantly, we uncovered trialkylamines as a cheap and readily available alternative to the previously reported electron–proton donor system consisting of Hantzsch ester/Brønsted acid. By employing the trialkylamine/photocatalyst system, we have been able to develop an efficient intramolecular ketyl–olefin and ketyl–alkyne coupling employing readily prepared substrates. The formal hydroacylation protocol provides the chromanol derivatives in good yield under mild reaction conditions and with low catalyst loadings. Efforts are currently underway to expand this concept to further transformations.
8. Nickel-Catalyzed Csp2–Csp3 Cross-Coupling via C–O Bond Activation
Lin Guo, Chien-Chi Hsiao, Huifeng Yue, Xiangqian Liu, Magnus Rueping
ACS Catal. 2016, 6, 4438-4442

Abstract:
We describe a new and practical way to construct Csp2–Csp3 bonds by using B-alkyl-9-BBNs as a coupling partner in nickel-catalyzed C–O bond activation. Given the widespread utility of catalytic alkylations in academic and industrial settings, the results provide new application opportunities as the reactions avoid the use of halides and sulfonates and allow displacement of the pivalate group, which can be used as a directing group in C–H functionalization prior to the dealkoxygenative alkylations. This new protocol also exhibits good compatibility with a variety of synthetically important functional groups of phenol pivalates, as well as alkylboranes. To further demonstrate the utility of this method, a new strategy for sequential functionalization of aromatic rings was performed by selective and sequential C–O bond activation.
Eleonora Fava, Anthony Millet, Masaki Nakajima, Sebastian Loescher, Magnus Rueping
Angew. Chem. Int. Ed. 2016, 55, 6776-6779

Abstract:
We report a direct photocatalytic method for the synthesis of unsymmetric 1,2-diamines, with reductive SET umpolung of an aldimine as the key step. Moreover, we also applied the concept of reductive umpolung to aldehyde–aniline couplings. Ketyl radicals derived from aldehydes were thus coupled in an intermolecular fashion by photoredox catalysis for the first time. Biologically active amino alcohols and 1,2-diamines can thus be directly synthesized from simple starting materials in a straightforward library format. Remarkably, a wide range of functional groups as well as amino acids were tolerated under the mild conditions of our photoredox process.
Xiangqian Liu, Chien‐Chi Hsiao, Indrek Kalvet, Matthias Leiendecker, Lin Guo, Franziska Schoenebeck, Magnus Rueping
Angew. Chem. Int. Ed. 2016, 55, 6093-6098
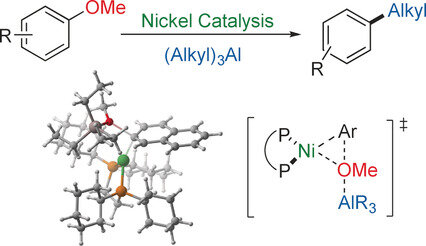
Abstract:
A newly developed Lewis acid assisted nickel-catalyzed cross-coupling reaction constitutes the first broadly applicable method for the alkylation of anisole derivatives under C−OMe bond cleavage. This was achieved as a result of the effective interplay of various factors: the Lewis acidic trialkylaluminums facilitate the oxidative addition by activating the C−OMe bond, the formation of stable dialkylaluminum methoxide favors an efficient transmetalation step, and the nickel catalyst with a bidentate dcype ligand undergoes the catalytic cycle for the tested substrates efficiently, suppressing the competing β-hydride elimination and affording the desired products with high yields. Naturally occurring and synthetically easily accessible structurally diverse anisole derivatives can thus be efficiently transformed into a variety of alkyl-substituted molecules. Building on our newly developed Lewis acid assisted cross-coupling reaction, we further demonstrated diversity-oriented, anisole-based strategies for the synthesis of a variety of novel products.
Adisak Chatupheeraphat, Hsuan‐Hung Liao, Steffen Mader, Makoto Sako, Hiroaki Sasai, Iuliana Atodiresei, Magnus Rueping
Angew. Chem. Int. Ed. 2016, 55, 4803-4807

Abstract:
We report a novel Brønsted acid catalyzed enantioselective sulfa- and oxa-Michael addition to in situ generated quinone methides. The generation of the intermediate aza-ortho-quinone methide proceeds under very mild reaction conditions in the presence of low amounts of chiral phosphoric acids and leads to the corresponding benzylic ethers and thioethers with excellent yields and enantioselectivities. As functionalized diaryl methanol ethers and the corresponding thioethers are difficult to access, the herein presented asymmetric organocatalytic addition reaction should be of great benefit in the synthesis of biologically relevant compounds.
Pavlo Nikolaienko, Magnus Rueping
Chem. Eur. J. 2016, 22, 2620-2623

Abstract:
A mild procedure for the trifluoromethylselenolation of aromatic compounds was developed. Various readily available aryl diazonium tetrafluoroborates smoothly underwent reactions to give products in good to high yields. Both electron-rich and -poor substrates are suitable for the process, and different functional groups, such as nitro, halogen, ether, ester, amino, nitrile, and carbonyl, are tolerated under the reaction conditions. The scope of the reaction was extended by the utilization of pentafluoroethyltrimethylsilane and various heteroaryldiazonium tetrafluoroborates. In addition, a one-pot approach starting from the corresponding anilines provided the trifluoroselenolated products with good overall yield on a gram-scale.
Pavlo Nikolaienko, Tülay Yildiz, Magnus Rueping
Eur. J. Org. Chem. 2016, 2016, 1091-1094

Abstract:
A new method for the Cu-mediated C–SCF3 bond-forming reaction is described. Extensive investigations showed that no ligand and additives are necessary. By applying this mild and rapid protocol, a variety of diaryliodonium salts were converted into their corresponding aryl trifluoromethyl thioethers in good yields Moreover, the scope was widened by utilizing heteroaromatic iodonium salts, and many of the presented (heteroaryl)(mesityl)iodonium salts were unknown prior this work. Due to the wide spectrum of biological activities exhibited by Ar–SCF3 compounds, mild reaction conditions and the simplicity of our protocol, we expect this method to be added to the range of synthetic approaches affording aryl trifluoromethyl sulfides.
Eleonora Fava, Masaki Nakajima, Martin B. Tabaka, Magnus Rueping
Green Chem. 2016, 18, 4531-4535

Abstract:
We have developed a mild and tin-free method for the preparation of synthetically valuable γ-lactams in good yields and high diastereomeric ratios, using visible light photoredox catalysis. Therewith, we provide the first example of visible light mediated 5-endo-trig cyclisation of α-chloroenamides that, in spite of its synthetic utility, still remains a challenging transformation. Furthermore, the diastereoselective reaction resulted in the formation of a single diastereoisomer, which is of interest for the synthesis of enantiomerically enriched products. The use of α-chloroenamides for the generation of monocyclic γ-lactams is particularly appealing and further application of this photoredox catalysis methodology is currently under investigation.
Quentin Lefebvre, Norbert Hoffmann, Magnus Rueping
Chem. Commun. 2016, 52, 2493-2496

Abstract:
The first photoorganocatalytic trifluoromethylation of electron-deficient olefins using a very simple and inexpensive benzophenone derivative as organic photocatalyst is described. These conditions could be directly used without further optimisation for the trifluoromethylation of (hetero)aromatics, thus giving an unified method for the direct trifluoromethylation of organic molecules using sodium triflinate as a cheap and safe alternative to other CF3 radical sources such as CF3I or CF3SO2Cl. The use of a photo-flow setup led to dramatically decreased reactions times, which shows the potential of our method for rapid scale-up and in-line synthesis. We also demonstrated that this transformation proceeds with similar or even higher yields using visible light and an iridium-based photocatalyst. Thus, the new methodology will find applications in drug discovery and in the synthesis of complex fluorinated molecules