
Excited-state palladium-catalyzed defluorinative arylation of polyfluoroarenes
Arnab Dey, Cătălin C. Anghel, Rajesh Kancherla, Magnus Rueping
Chem. Commun. 2026, Advance Article
DOI: 10.1039/d6cc03082c
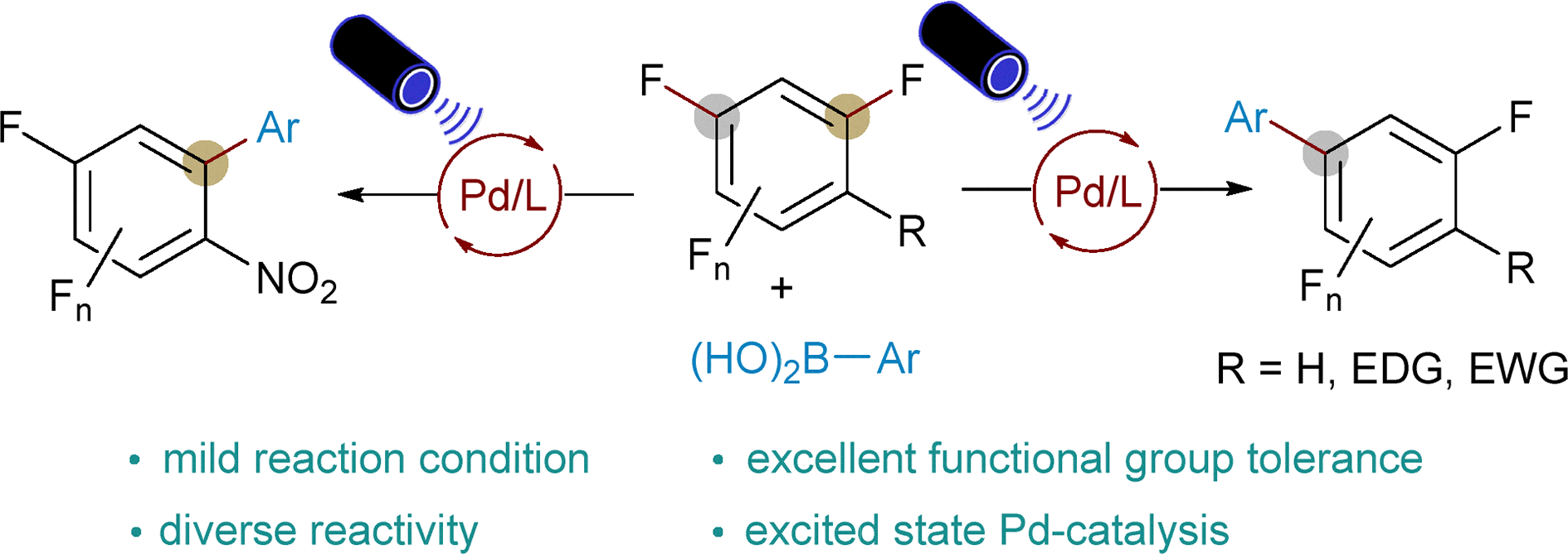
Abstract:
Visible-light-induced excited-state palladium catalysis enables defluorinative C(sp2)–C(sp2) cross-coupling of polyfluoroarenes with aryl boronic acids under blue LED irradiation at room temperature. The Pd(OAc)2/BrettPhos catalyst selectively activates C(sp2)–F bonds without external photocatalysts, affording multifluorinated biaryls in good to excellent yields through photoinduced formation of Ar–Pd(ii) intermediates.
Photoinduced Palladium-Catalyzed Site-Selective Meta-Alkylation of Pyridines
Anurag Singh, Rajesh Kancherla, Sayan Dutta, Arnab Dey, Kuntal Pal, Bholanath Maity, Lennard Kloene, Luigi Cavallo, Rene M. Koenigs, Magnus Rueping
Angew. Chem. Int. Ed. 2026, e5426146.
DOI: 10.1002/anie.5426146
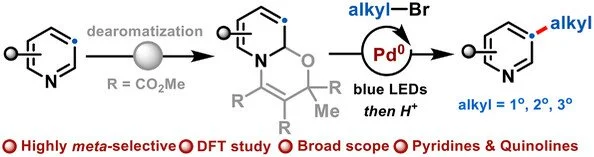
Abstract:
Achieving regioselective meta-C–H alkylation of pyridines remains a long-standing challenge due to their intrinsic electronic bias and the lack of general strategies compatible with simple alkyl electrophiles. In contrast to Minisci-type alkylations, which preferentially functionalize electron-deficient positions, meta-alkylation of unbiased pyridines using alkyl bromides has remained elusive. Herein, we report a photoinduced palladium-catalyzed strategy that enables regioselective meta-alkylation of pyridines with commercially available alkyl bromides via a redox-neutral dearomatization pathway. Visible-light excitation of palladium generates a hybrid alkyl–Pd(I) radical species that undergoes regioselective addition to oxazino-pyridine intermediates despite inherent polarity mismatch, forming a stabilized π-allyl Pd(I) radical prior to rearomatization. The method exhibits a broad substrate scope encompassing diverse pyridines, quinolines, and alkyl bromides, and enables one-pot and sequential meta-functionalization strategies. DFT calculations support an outer-sphere radical addition mechanism and rationalize the observed regioselectivity. This work establishes a new paradigm for Pd-catalyzed meta-C–H alkylation of azines, a complementary conceptual approach to conventional and important Minisci chemistry.
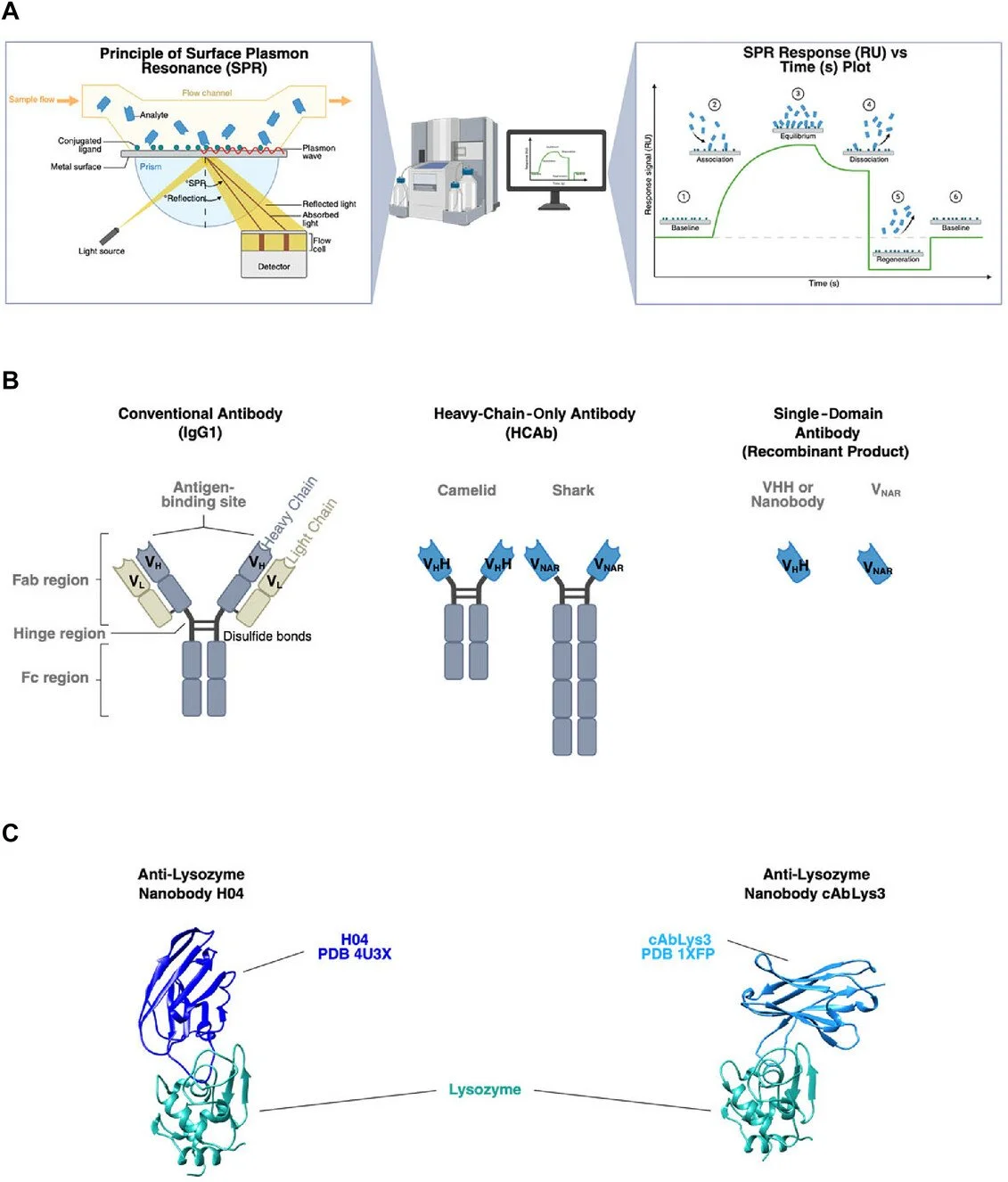
Escarlet Díaz-Galicia, Nicoleta Gutu, Yuli Peng, Almira Valitova, Dominik Renn, Magnus Rueping
Current Protocols, 2026, 6, e70360
DOI: 10.1002/cpz1.70360
Abstract:
Developing protein interaction–based technologies such as biosensors requires a clear understanding of receptor-target kinetics. Nanobodies, which are camelid-derived single-domain antibodies, are ideal biosensor receptors due to their high specificity, stability, and ease of production. During biosensor development, multiple nanobody variants are often tested against the same target to identify the best binders. Surface plasmon resonance (SPR) is a robust, label-free method for measuring these interactions, but its many experimental variables can complicate the establishment of a streamlined protocol specially for non-high-throughput instruments. Here, we present an SPR workflow that enables the comparative analysis of nanobody variants binding to lysozyme as a model target on a Biacore T100 instrument. These protocols cover the steps from protein expression and purification to final affinity ranking.
Pulsed electrolysis enables unexpected lactonization of bicyclobutane carboxylic acids
Anton S. Makarov, Liang Yi, Bholanath Maity, Luigi Cavallo, Magnus Rueping
Chem. Sci., 2026, Advance Article
DOI: 10.1039/D6SC01541G
Abstract:
Anodic oxidation of bicyclobutane carboxylic acids leads to unexpected lactonization and provides access to substituted benzene bioisosteres from highly strained precursors. High selectivity was achieved under pulsed electrolysis. Mechanistic experiments, supported by computational analysis, indicate that the transformation is initiated by single-electron oxidation of the substrate to form a bicyclobutyl radical cation, a key intermediate in redox-mediated transformations. These results demonstrate the utility of electrochemical methods for controlling the reactivity of bicyclobutane derivatives and provide a foundation for further development of sustainable oxidative transformations of strained molecules.
Photochemical, electrochemical and electrophotochemical C−N bond-forming cross-coupling reactions
Bo Li, Nian Li, Huabin Zhang, Wen-Jing Xiao, Magnus Rueping
Nat Rev Chem, 2026, 10, 332–349
DOI: 10.1038/s41570-026-00819-6
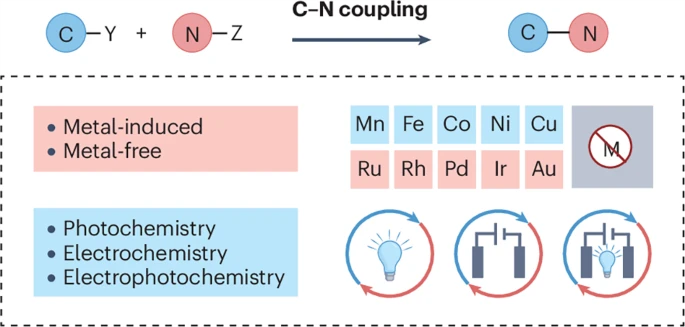
Abstract:
Nitrogen-containing molecules are foundational to modern synthesis, spanning a wide range of pharmaceuticals, agrochemicals, life science and functional materials. Recent advances in photochemical and electrochemical methods have begun to redefine how C–N bonds can be constructed, expanding beyond traditional thermally driven metal-catalysed cross-couplings to encompass radical, hybrid, and fully metal-free pathways. These strategies can offer precise control over redox events, access to otherwise inaccessible intermediates and greater modularity in reaction design, thereby enabling transformations that are operationally simple, scalable and increasingly sustainable. In this Review, we systematically summarize photochemical, electrochemical and electrophotochemical C–N cross-coupling reactions, spanning mechanistic principles, catalytic paradigms, reaction classes and synthetic applications. We highlight convergent trends across these fields, analyse their distinct advantages and provide a forward-looking perspective on their potential and future development.
Dominik Renn, Justine S. McPartlan, Srinivas Banala, Fabian Kiessling, Poulami Talukder, Christian W. Mandl, Jörg Eppinger, Jasdave S. Chahal, Magnus Rueping
Sci Rep, 2026, 16, 14565
DOI: 10.1038/s41598-026-44645-8
Abstract:
The continuing emergence of SARS- and MERS-related coronaviruses underscores the urgent need for pan-SARBECo vaccines capable of eliciting broad and durable protective immune responses across divergent lineages (Cankat et al. in Cell. Mol. Immunol. 21(2):103–118, 2024). We present a heterologous prime-boost vaccination strategy combining a modified dendrimer nanoparticle (DNP)-encapsulated self-amplifying (saRNA) prime with an alum-adjuvanted multivalent protein booster containing receptor-binding domains (RBDs) from SARS-CoV-2 (Wuhan-Hu-1 and B.1.351) and MERS-CoV. This approach leverages the potent immunogenicity of RNA priming together with the breadth and safety of protein subunit boosting (Bruno et al. in Npj Vaccines 10(1):108, 2025; Kim et al. in Vaccines 13(8):797, 2025) to expand coronavirus coverage. In preclinical mouse and hamster models, the heterologous RNA-protein regimen elicited robust antibody responses with markedly enhanced magnitude, durability, and cross-variant breadth compared with homologous RNA or protein vaccination alone. Inclusion of the MERS-CoV RBD in the booster broadened the response without compromising SARS-CoV-2 immunity. These findings establish a versatile and scalable vaccination strategy with potential to inform the development of next-generation, broadly protective vaccines against emerging coronaviruses.
Chao Wan, Chao Yang, Magnus Rueping, Chen Zhu, Lin Guo, Wujiong Xia
Nat. Chem. 2026, 18, 386–397
DOI: 10.1038/s41557-025-02001-9
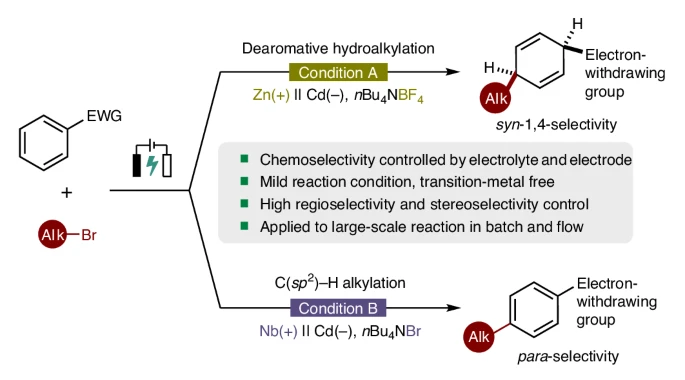
Abstract:
Dearomative functionalization of arenes represents a powerful synthetic strategy for the rapid assembly of complex chemical architectures. A significant challenge in this process is overcoming the inherent aromaticity of arenes. Here, leveraging the potential of organic electrolysis, we show the development of a dearomative syn-1,4-hydroalkylation reaction targeting electron-deficient arenes and heteroarenes. This electrochemical approach, conducted under mild, operationally straightforward and scalable conditions, facilitates the synthesis of alkylated syn-1,4-cyclohexadienes with high chemoselectivity, regioselectivity and stereoselectivity. In addition, this alkylation protocol is controllable and switchable. By employing a niobium plate as the anode and nBu4NBr as the supporting electrolyte, our method enables the para-selective C(sp2)–H alkylation of (hetero)arenes via electrolysis. Both reactions exhibit broad substrate scope and demonstrate excellent compatibility with various electron-deficient arenes and alkyl bromides. Furthermore, preliminary mechanistic studies and density functional theory calculations have been performed to elucidate the reaction mechanism and to rationalize the observed chemoselectivity, regioselectivity and stereoselectivity.
Predictive, Data-Driven Design of Red-Light Photoredox Catalysts for C─Heteroatom Bond Formation
Amir Gizatullin, Tingting Yuan, Sascha Grotjahn, Luigi Cavallo, Burkhard König, Chen Zhu, Magnus Rueping
Angew. Chem. Int. Ed. 2026, e26086
DOI: 10.1002/anie.202526086
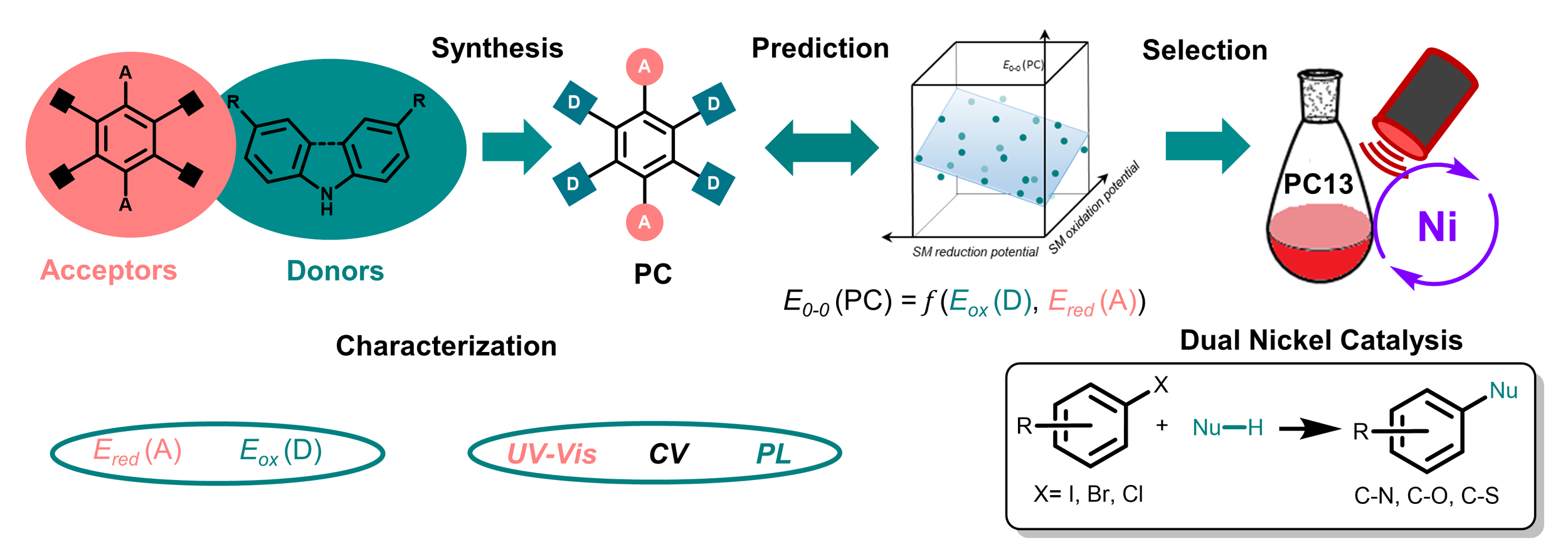
Abstract:
Photocatalysis is a powerful tool for the synthesis of organic molecules, yet its widespread application is hindered by the dependence on high-energy light sources and expensive metal-based catalysts, which can limit scalability and environmental sustainability. In this study, we present a modular design strategy for organic dyes engineered for efficient red-light absorption, enabling photocatalytic reactions under low-energy irradiation. Our findings establish a clear relationship between the oxidation potential of the photocatalyst and the nature of its donor moiety, as well as between the reduction potential and the electronic characteristics of its core structure. Moreover, we demonstrate that the E0-0 energy of a photocatalyst can be predicted via multivariate linear regression using the donor's oxidation potential and the core's reduction potential as descriptors. Utilizing this strategy, we synthesized red-light-absorbing photocatalysts that efficiently promote C─heteroatom cross-coupling reactions under mild conditions. This approach overcomes the limitations of blue-light photocatalysis by offering broad substrate compatibility, including π-conjugated aryl bromides and photolabile functional groups, while minimizing undesirable hydrodehalogenation. By reducing reliance on precious metals and improving energy efficiency, our approach provides a scalable alternative to traditional photocatalysis and advances the development of metal-free photocatalysts for sustainable chemistry.
Prashant S. Shinde, Valmik S. Shinde, Magnus Rueping
Chem. Sci., 2026, 17, 652-663
DOI: 10.1039/D5SC05919D
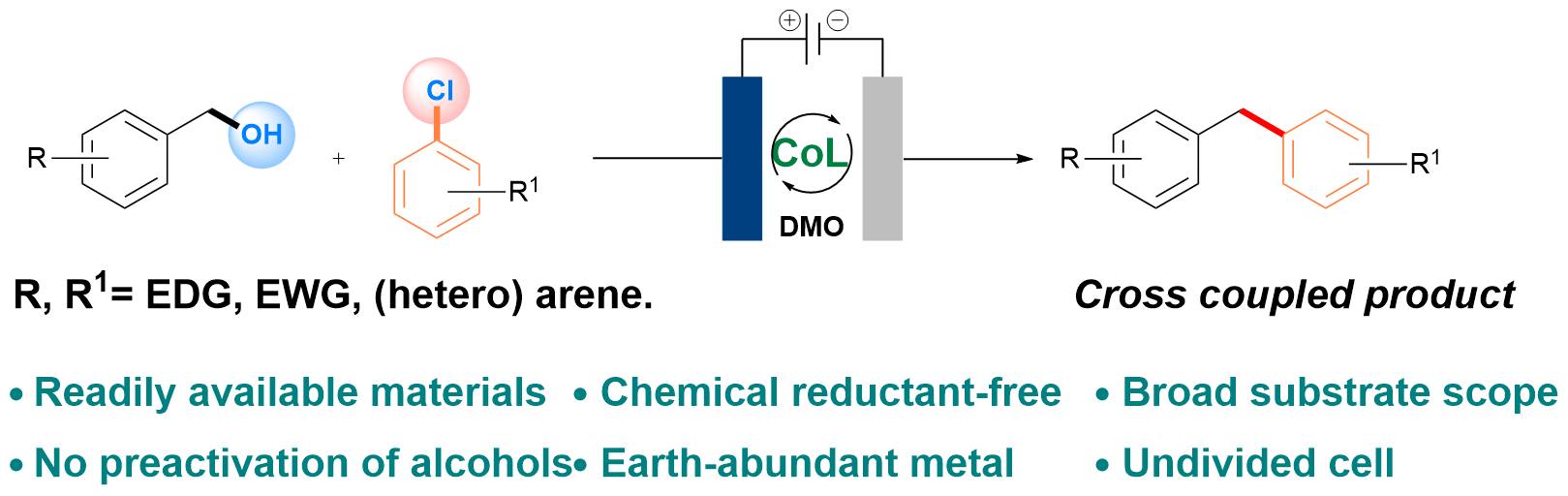
Abstract:
The direct functionalization of alcohols via C–O bond cleavage is a synthetically valuable but challenging transformation. In this work, we report a reductive C(sp3)–C(sp2) cross-coupling reaction between benzyl alcohols and a broad range of aryl chlorides. The success of this transformation is attributed to the development of low-coordinate cobalt/bipyridine complexes, which enable the selective conversion of benzyl alcohols into the corresponding diarylmethanes, while minimizing undesired homocoupling of either the benzyl alcohol or aryl chlorides.
Moyu Yi, Karthik Peramaiah, Xingzhu Chen, Renqian Zhou, Nursaya Zhumabay, Hongbin Dou, Hao Huang , Indranil Dutta, Shouwei Zuo, Jifeng Wu, Bin Chang, Tsu-Chien Weng, Magnus Rueping, Osman M. Bakr, Huabin Zhang, Kuo-Wei Huang
Appl. Catal. B: Environ., 2026, 383, 126126
DOI: 10.1016/j.apcatb.2025.126126
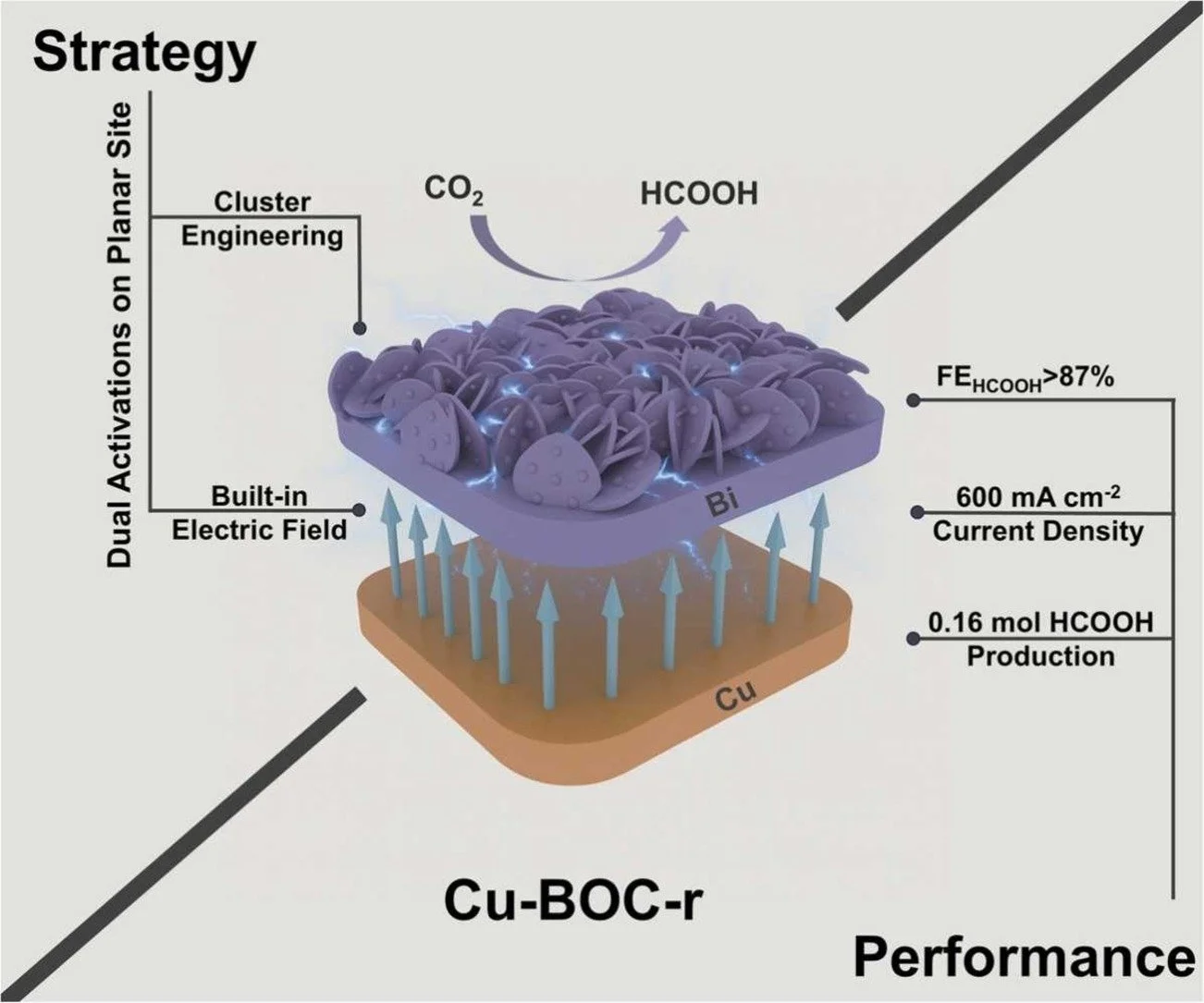
Abstract:
Activating catalytically inert planar sites and achieving high-rate CO2 to formic acid conversion remain key challenges for industrial implementation of bismuth nanoflower-based electrocatalyst. Here, we integrate in-situ reconstruction of Bi nanoclusters with heterointerfacial electric fields at a Bi/Cu junction. The dual (geometric + electronic) activation creates secondary cluster sites, enhances interfacial charge transformation, and improves the selectivity to the key product formic acid. The resulting catalyst achieves > 87 % Faradaic efficiency across a wide current density range (100–600 mA cm−2) and enables mole-scale production of pure formic acid (≈ 0.16 mol) over 100 h of continuous operation in a solid electrolyte reactor. In-situ studies and DFT calculations reveal a synergistic mechanism of planar surface activation by cluster engineering and electric field-enhanced intermediate binding. This work establishes a scalable and integrative catalyst-interface-reactor framework for practical CO2 electrolysis toward a liquid hydrogen carrier.
Resonant acoustic mixing enables solvent-less amide coupling in solid-phase peptide synthesis
Alice Nanni, Panayiotis Bilalis, Magnus Rueping
Green Chem., 2026, 28, 255-263
DOI: 10.1039/D5GC04067A

Abstract:
Solid-phase peptide synthesis (SPPS) is the backbone of modern peptide production. However, it relies heavily on relatively toxic solvents and generates significant waste, limiting its sustainability and scalability. To address these limitations, we report the first fully solvent-less peptide coupling protocol for SPPS enabled by Resonant Acoustic Mixing (RAM), representing a step toward greener peptide manufacturing. This method eliminates bulk solvent use, reagent pre-dissolution, and pre-activation during coupling by using mechanical agitation to drive efficient amide bond formation. Optimized conditions (95g acceleration, 5 min, 1.5 equiv. Fmoc-amino acids) afford rapid and clear reactions with high conversion and purity. Notably, no external solvent is added during coupling; instead, residual solvent retained from resin pre-swelling creates a localized microenvironment sufficient for in situ activation. Compared to conventional SPPS, this protocol significantly reduces solvent and reagent use, reaction time, and waste. Process Mass Intensity (PMI) calculations show clear improvements, highlighting the method's environmental and economic benefits. This approach was validated by synthesizing two bioactive peptides (IKVAV and Angiotensin 1–7) in high yield and purity, and further demonstrated excellent scalability in a tenfold scale-up.